Mathematical Biology Section, Laboratory of Biological Modeling, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, South Drive, Bethesda, MD 20814, USA.
Mathematical Biology Section, Laboratory of Biological Modeling, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, South Drive, Bethesda, MD 20814, USA ; Department of Psychology, University of Minnesota Twin Cities, 75 East River Parkway, Minneapolis, MN 55455, USA ; Cognitive Genomics Lab, BGI Shenzhen, Yantian District, Shenzhen, China.
Gigascience. 2014 Jun 16;3:10. doi: 10.1186/2047-217X-3-10. eCollection 2014.
The aim of a genome-wide association study (GWAS) is to isolate DNA markers for variants affecting phenotypes of interest. This is constrained by the fact that the number of markers often far exceeds the number of samples. Compressed sensing (CS) is a body of theory regarding signal recovery when the number of predictor variables (i.e., genotyped markers) exceeds the sample size. Its applicability to GWAS has not been investigated.
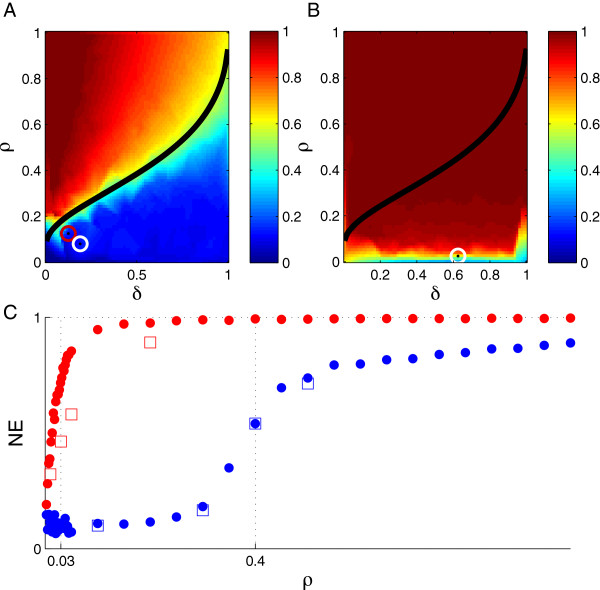
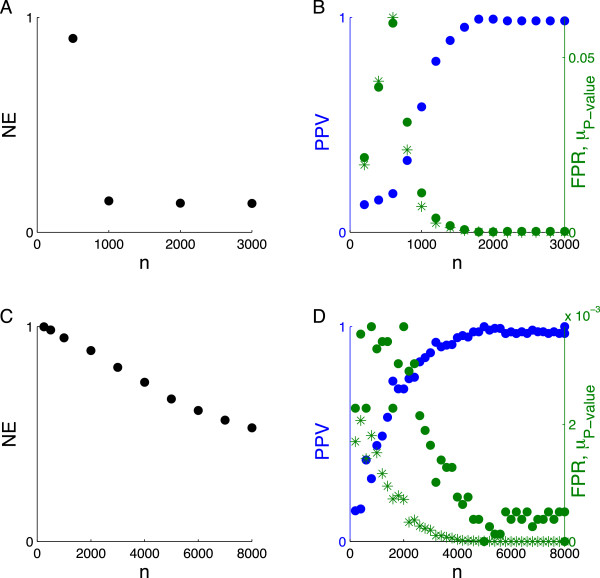
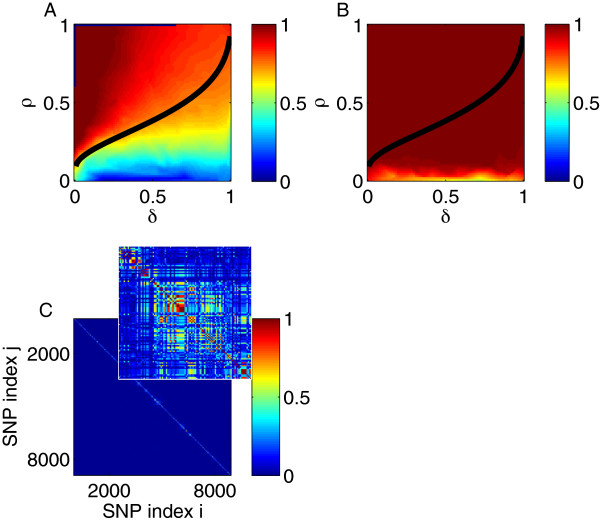
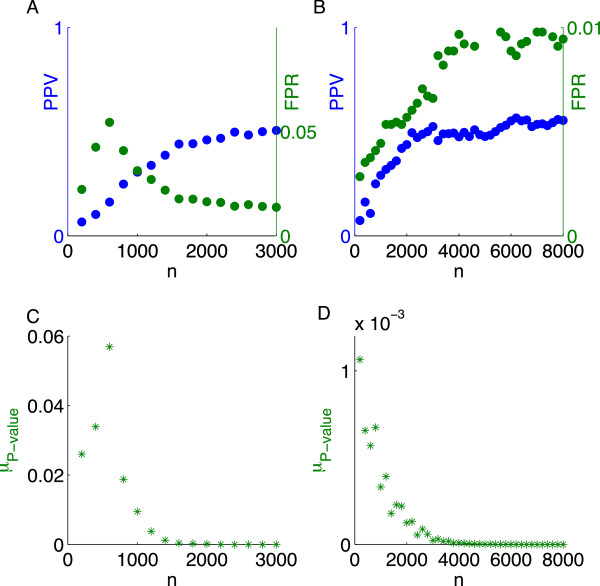
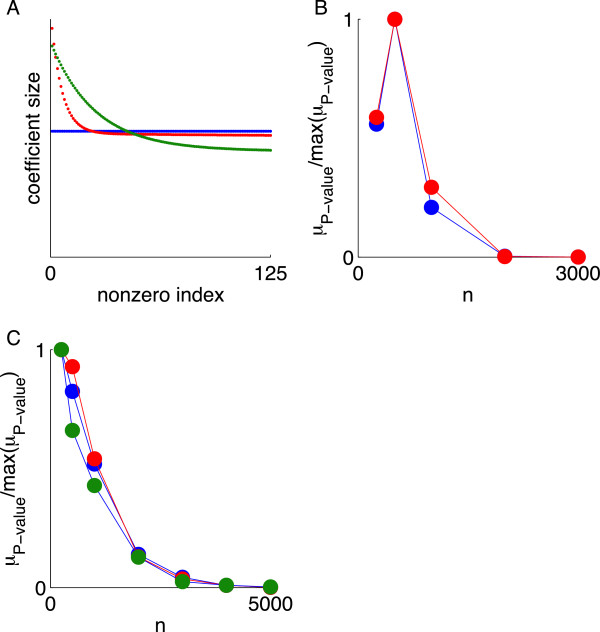
Using CS theory, we show that all markers with nonzero coefficients can be identified (selected) using an efficient algorithm, provided that they are sufficiently few in number (sparse) relative to sample size. For heritability equal to one (h (2) = 1), there is a sharp phase transition from poor performance to complete selection as the sample size is increased. For heritability below one, complete selection still occurs, but the transition is smoothed. We find for h (2) ∼ 0.5 that a sample size of approximately thirty times the number of markers with nonzero coefficients is sufficient for full selection. This boundary is only weakly dependent on the number of genotyped markers.
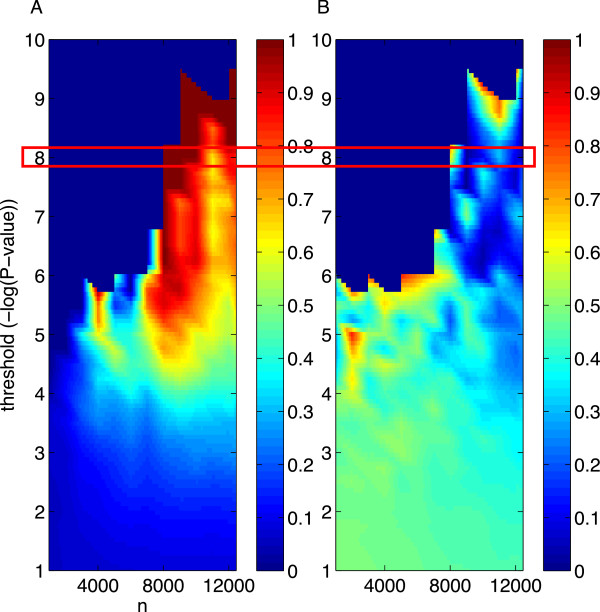
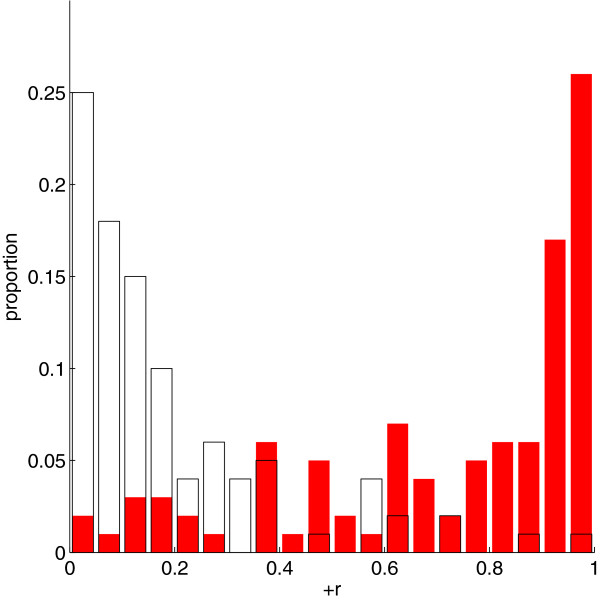
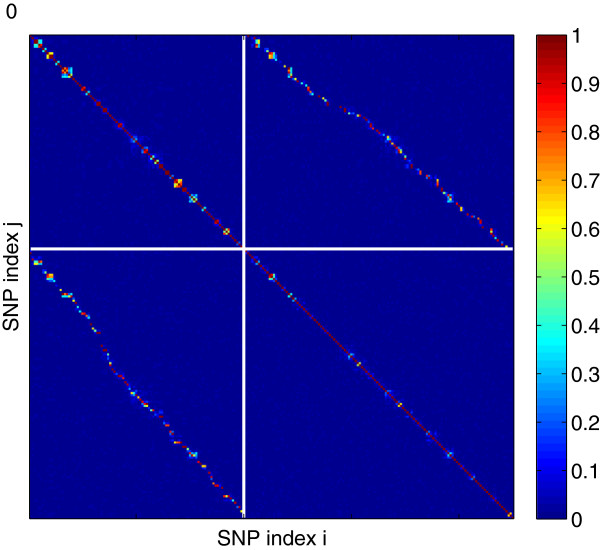
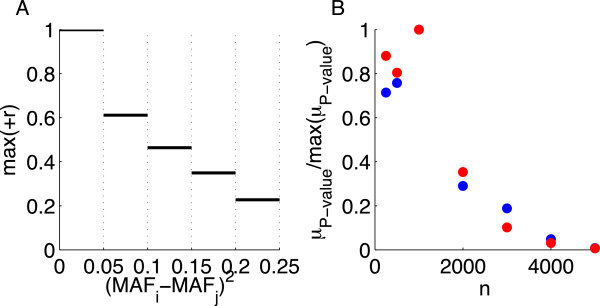
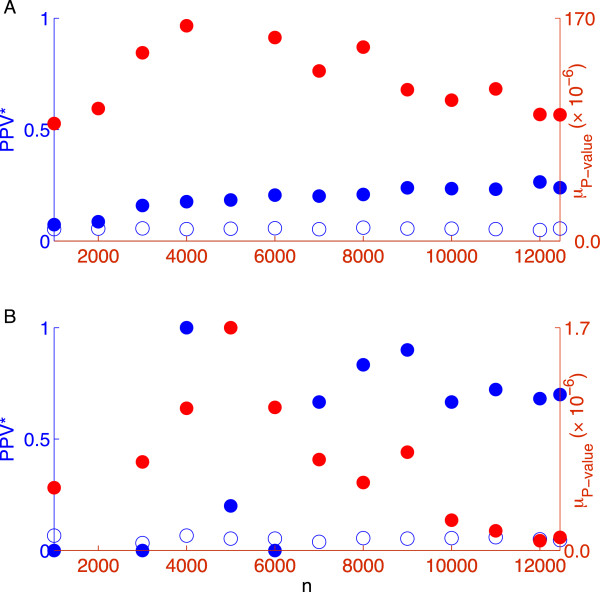
Practical measures of signal recovery are robust to linkage disequilibrium between a true causal variant and markers residing in the same genomic region. Given a limited sample size, it is possible to discover a phase transition by increasing the penalization; in this case a subset of the support may be recovered. Applying this approach to the GWAS analysis of height, we show that 70-100% of the selected markers are strongly correlated with height-associated markers identified by the GIANT Consortium.
全基因组关联研究(GWAS)的目的是分离影响感兴趣表型的 DNA 标记变体。这受到这样一个事实的限制,即标记的数量通常远远超过样本的数量。压缩感知(CS)是一种关于预测变量(即,基因分型标记)数量超过样本量时信号恢复的理论。它在 GWAS 中的适用性尚未得到研究。
使用 CS 理论,我们表明,只要它们的数量相对于样本量足够少(稀疏),则可以使用有效的算法识别(选择)所有具有非零系数的标记。对于遗传率等于一(h(2)= 1),随着样本量的增加,从性能不佳到完全选择会出现明显的相变。对于遗传率低于一,仍然会发生完全选择,但相变被平滑化。我们发现对于 h(2)≈0.5,大约是具有非零系数的标记数量的三十倍的样本量足以进行完全选择。该边界仅与基因分型标记的数量弱相关。
实际的信号恢复措施对于真实因果变体与位于同一基因组区域中的标记之间的连锁不平衡具有鲁棒性。给定有限的样本量,通过增加惩罚可以发现相变;在这种情况下,可能会恢复支持的子集。将此方法应用于身高的 GWAS 分析,我们表明,选择的标记中有 70-100%与 GIANT 联盟确定的与身高相关的标记强烈相关。