Department of Physics and Astronomy, Michigan State University, East Lansing, USA.
Genomic Prediction, North Brunswick, NJ, USA.
Sci Rep. 2020 Jul 21;10(1):12055. doi: 10.1038/s41598-020-68881-8.
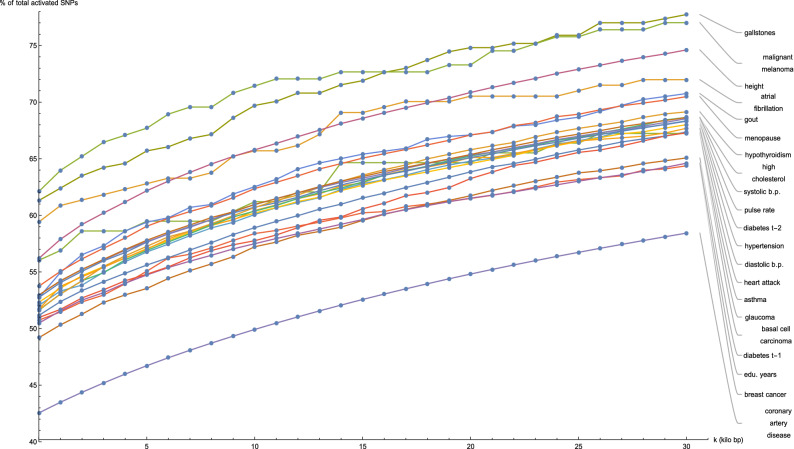
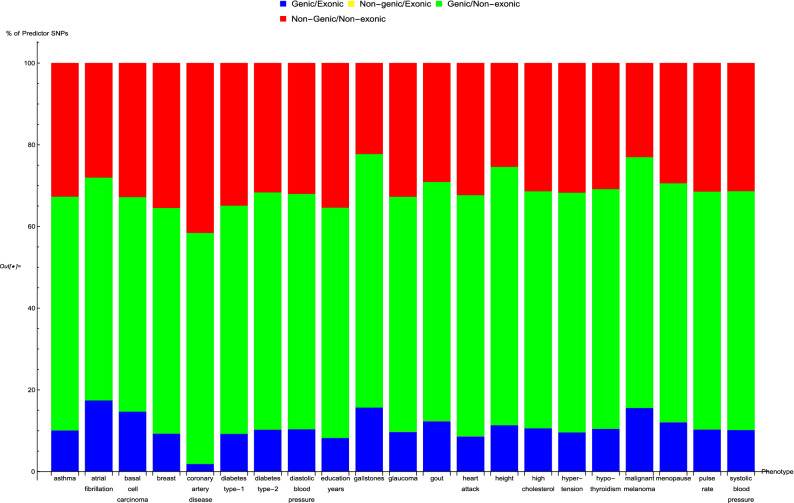
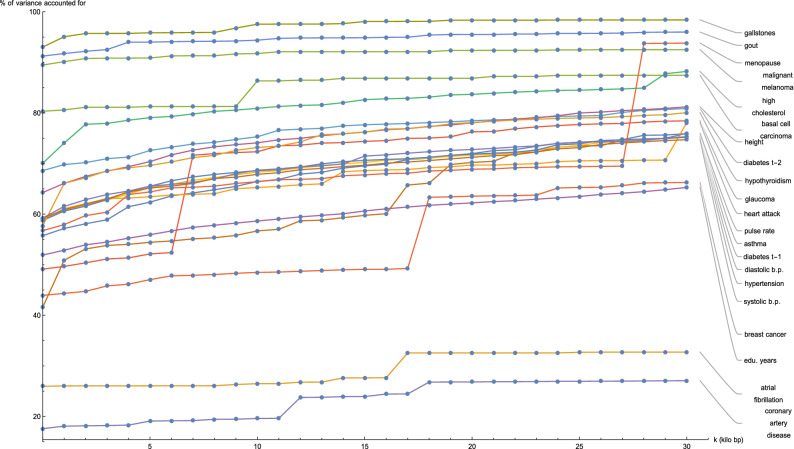
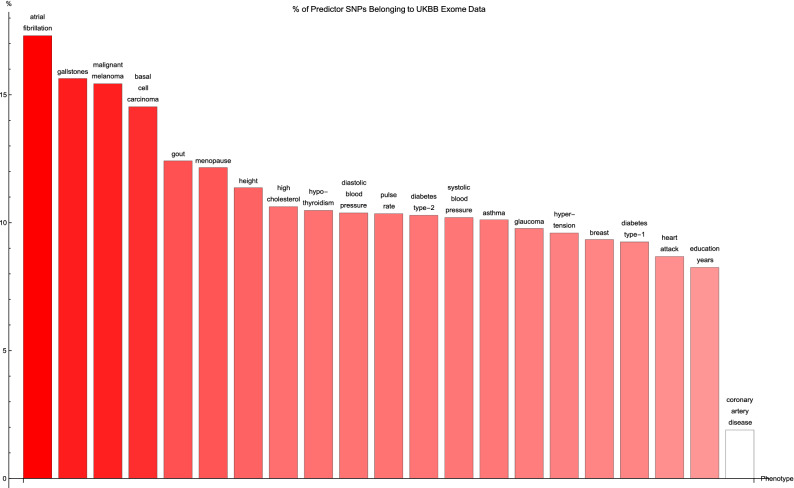
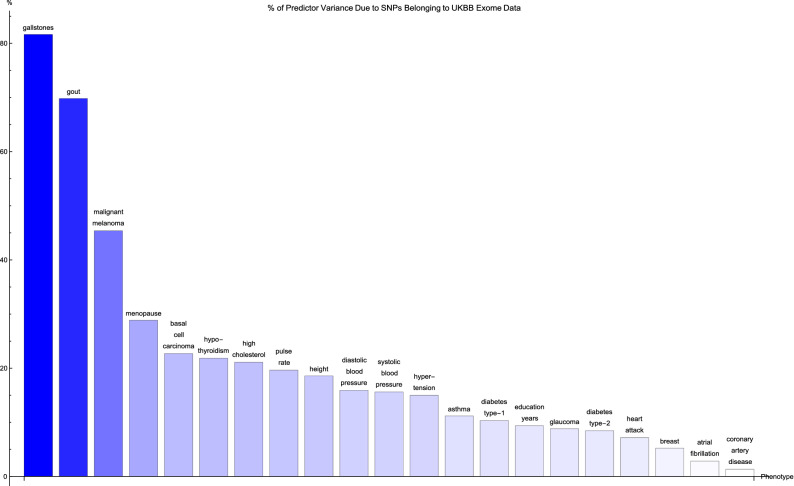
Genomic prediction of complex human traits (e.g., height, cognitive ability, bone density) and disease risks (e.g., breast cancer, diabetes, heart disease, atrial fibrillation) has advanced considerably in recent years. Using data from the UK Biobank, predictors have been constructed using penalized algorithms that favor sparsity: i.e., which use as few genetic variants as possible. We analyze the specific genetic variants (SNPs) utilized in these predictors, which can vary from dozens to as many as thirty thousand. We find that the fraction of SNPs in or near genic regions varies widely by phenotype. For the majority of disease conditions studied, a large amount of the variance is accounted for by SNPs outside of coding regions. The state of these SNPs cannot be determined from exome-sequencing data. This suggests that exome data alone will miss much of the heritability for these traits-i.e., existing PRS cannot be computed from exome data alone. We also study the fraction of SNPs and of variance that is in common between pairs of predictors. The DNA regions used in disease risk predictors so far constructed seem to be largely disjoint (with a few interesting exceptions), suggesting that individual genetic disease risks are largely uncorrelated. It seems possible in theory for an individual to be a low-risk outlier in all conditions simultaneously.
近年来,复杂人类特征(例如身高、认知能力、骨密度)和疾病风险(例如乳腺癌、糖尿病、心脏病、心房颤动)的基因组预测已经取得了相当大的进展。利用来自英国生物银行的数据,使用惩罚算法构建了预测因子,这些算法有利于稀疏性:即,使用尽可能少的遗传变异。我们分析了这些预测因子中使用的特定遗传变异(SNP),这些 SNP 可以从几十到多达三万不等。我们发现,表型中基因区域内或附近的 SNP 比例差异很大。对于大多数研究的疾病状况,大部分方差由编码区域之外的 SNP 解释。这些 SNP 的状态无法从外显子组测序数据中确定。这表明,仅外显子数据将错过这些特征的大部分遗传率,即,不能仅从外显子数据计算现有的 PRS。我们还研究了 SNP 比例和方差在两个预测因子之间的共同比例。到目前为止构建的疾病风险预测因子中使用的 DNA 区域似乎在很大程度上是不相交的(有几个有趣的例外),这表明个体遗传疾病风险在很大程度上是不相关的。从理论上讲,一个人在所有情况下同时成为低风险的异常值是有可能的。