BMC Genet. 2014 Aug 12;15:88. doi: 10.1186/s12863-014-0088-5.
Imputation of partially missing or unobserved genotypes is an indispensable tool for SNP data analyses. However, research and understanding of the impact of initial SNP-data quality control on imputation results is still limited. In this paper, we aim to evaluate the effect of different strategies of pre-imputation quality filtering on the performance of the widely used imputation algorithms MaCH and IMPUTE.
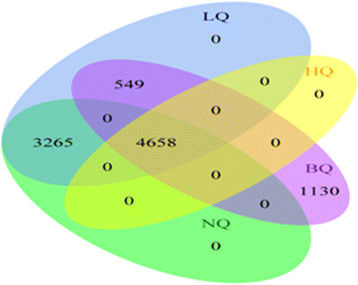
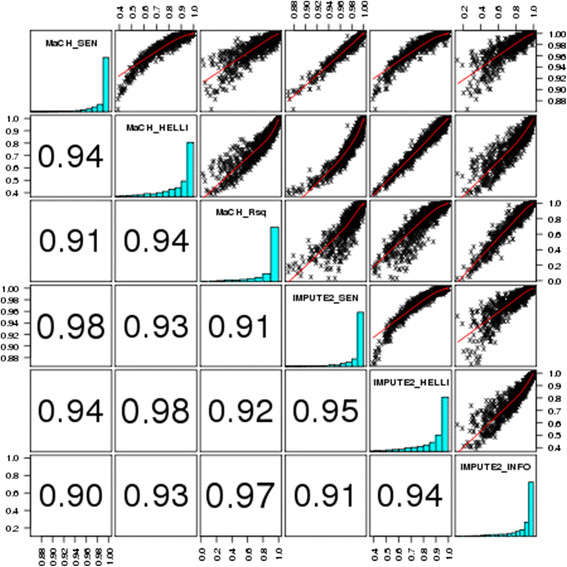
We considered three scenarios: imputation of partially missing genotypes with usage of an external reference panel, without usage of an external reference panel, as well as imputation of completely un-typed SNPs using an external reference panel. We first created various datasets applying different SNP quality filters and masking certain percentages of randomly selected high-quality SNPs. We imputed these SNPs and compared the results between the different filtering scenarios by using established and newly proposed measures of imputation quality. While the established measures assess certainty of imputation results, our newly proposed measures focus on the agreement with true genotypes. These measures showed that pre-imputation SNP-filtering might be detrimental regarding imputation quality. Moreover, the strongest drivers of imputation quality were in general the burden of missingness and the number of SNPs used for imputation. We also found that using a reference panel always improves imputation quality of partially missing genotypes. MaCH performed slightly better than IMPUTE2 in most of our scenarios. Again, these results were more pronounced when using our newly defined measures of imputation quality.
Even a moderate filtering has a detrimental effect on the imputation quality. Therefore little or no SNP filtering prior to imputation appears to be the best strategy for imputing small to moderately sized datasets. Our results also showed that for these datasets, MaCH performs slightly better than IMPUTE2 in most scenarios at the cost of increased computing time.
部分缺失或未观测基因型的插补是 SNP 数据分析不可或缺的工具。然而,对于初始 SNP 数据质量控制对插补结果的影响的研究和理解仍然有限。在本文中,我们旨在评估不同的预插补质量过滤策略对广泛使用的插补算法 MaCH 和 IMPUTE 的性能的影响。
我们考虑了三种情况:使用外部参考面板插补部分缺失基因型、不使用外部参考面板插补部分缺失基因型,以及使用外部参考面板插补完全未分型的 SNPs。我们首先创建了各种数据集,应用不同的 SNP 质量过滤并屏蔽一定比例的随机选择的高质量 SNPs。我们对这些 SNPs 进行了插补,并通过使用已建立和新提出的插补质量度量来比较不同过滤方案之间的结果。虽然已建立的度量评估插补结果的确定性,但我们新提出的度量侧重于与真实基因型的一致性。这些度量表明,预插补 SNP 过滤可能会对插补质量产生不利影响。此外,插补质量的最强驱动因素通常是缺失率和用于插补的 SNPs 数量。我们还发现,使用参考面板总是可以提高部分缺失基因型的插补质量。在我们的大多数场景中,MaCH 的表现略优于 IMPUTE2。同样,当使用我们新定义的插补质量度量时,这些结果更为明显。
即使是适度的过滤也会对插补质量产生不利影响。因此,在插补之前,对小数据集或中等大小的数据集进行少量或不进行 SNP 过滤似乎是最佳策略。我们的结果还表明,对于这些数据集,MaCH 在大多数情况下的表现略优于 IMPUTE2,但代价是计算时间增加。