Song Meng, Greenbaum Jonathan, Luttrell Joseph, Zhou Weihua, Wu Chong, Luo Zhe, Qiu Chuan, Zhao Lan Juan, Su Kuan-Jui, Tian Qing, Shen Hui, Hong Huixiao, Gong Ping, Shi Xinghua, Deng Hong-Wen, Zhang Chaoyang
School of Computing Sciences and Computer Engineering, University of Southern Mississippi, Hattiesburg, MS, United States.
Tulane Center of Biomedical Informatics and Genomics, School of Medicine, Tulane University, New Orleans, LA, United States.
Front Artif Intell. 2022 Nov 3;5:1028978. doi: 10.3389/frai.2022.1028978. eCollection 2022.
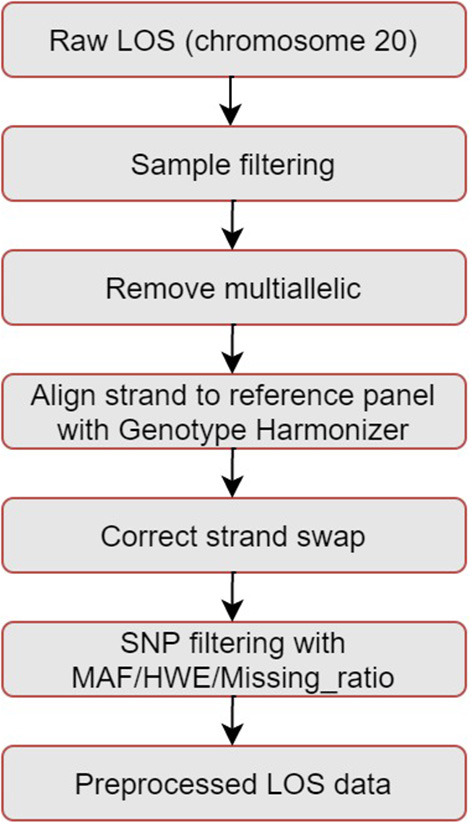
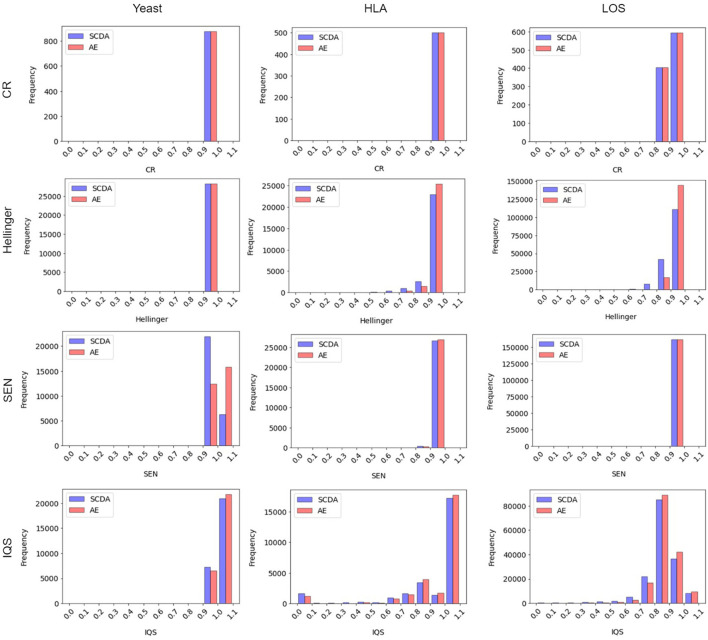
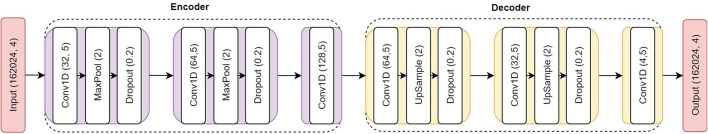
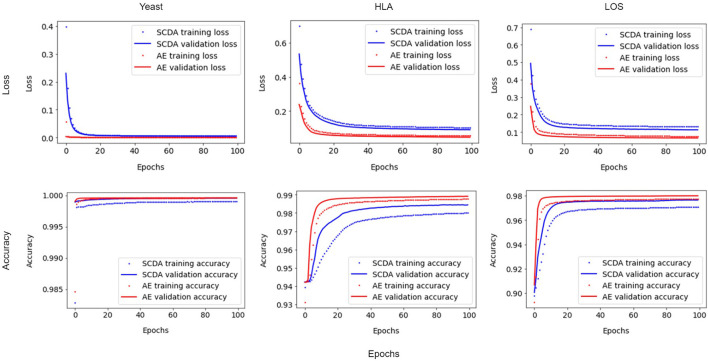
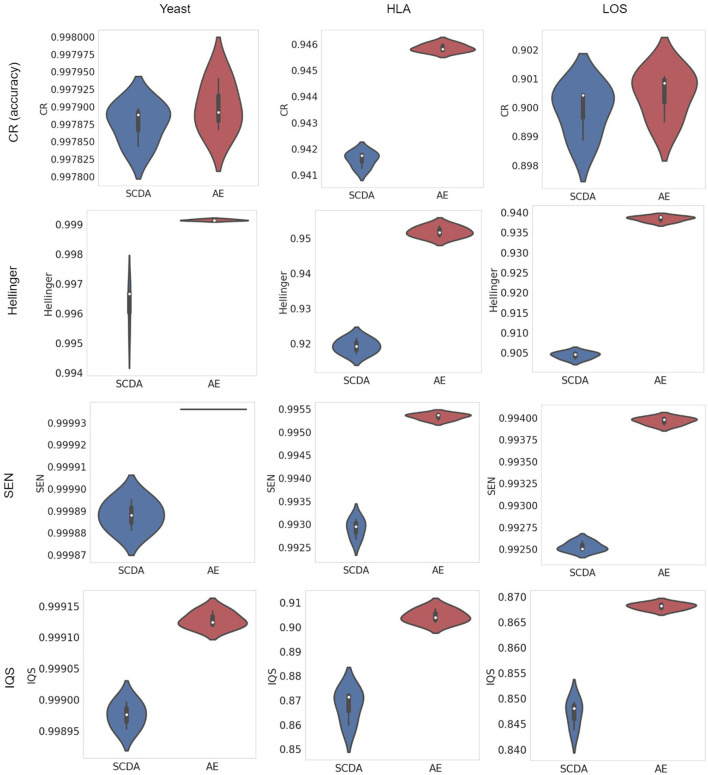
Genotype imputation has a wide range of applications in genome-wide association study (GWAS), including increasing the statistical power of association tests, discovering trait-associated loci in meta-analyses, and prioritizing causal variants with fine-mapping. In recent years, deep learning (DL) based methods, such as sparse convolutional denoising autoencoder (SCDA), have been developed for genotype imputation. However, it remains a challenging task to optimize the learning process in DL-based methods to achieve high imputation accuracy. To address this challenge, we have developed a convolutional autoencoder (AE) model for genotype imputation and implemented a customized training loop by modifying the training process with a single batch loss rather than the average loss over batches. This modified AE imputation model was evaluated using a yeast dataset, the human leukocyte antigen (HLA) data from the 1,000 Genomes Project (1KGP), and our in-house genotype data from the Louisiana Osteoporosis Study (LOS). Our modified AE imputation model has achieved comparable or better performance than the existing SCDA model in terms of evaluation metrics such as the concordance rate (CR), the Hellinger score, the scaled Euclidean norm (SEN) score, and the imputation quality score (IQS) in all three datasets. Taking the imputation results from the HLA data as an example, the AE model achieved an average CR of 0.9468 and 0.9459, Hellinger score of 0.9765 and 0.9518, SEN score of 0.9977 and 0.9953, and IQS of 0.9515 and 0.9044 at missing ratios of 10% and 20%, respectively. As for the results of LOS data, it achieved an average CR of 0.9005, Hellinger score of 0.9384, SEN score of 0.9940, and IQS of 0.8681 at the missing ratio of 20%. In summary, our proposed method for genotype imputation has a great potential to increase the statistical power of GWAS and improve downstream post-GWAS analyses.
基因型填充在全基因组关联研究(GWAS)中有广泛应用,包括提高关联检验的统计效能、在荟萃分析中发现与性状相关的基因座以及通过精细定位对因果变异进行优先级排序。近年来,已经开发了基于深度学习(DL)的方法,如稀疏卷积去噪自动编码器(SCDA)用于基因型填充。然而,在基于DL的方法中优化学习过程以实现高填充准确率仍然是一项具有挑战性的任务。为应对这一挑战,我们开发了一种用于基因型填充的卷积自动编码器(AE)模型,并通过使用单个批次损失而非批次平均损失来修改训练过程,实现了定制的训练循环。使用酵母数据集、来自千人基因组计划(1KGP)的人类白细胞抗原(HLA)数据以及我们来自路易斯安那骨质疏松症研究(LOS)的内部基因型数据对这种改进的AE填充模型进行了评估。在所有三个数据集中,就一致性率(CR)、海林格得分、缩放欧几里得范数(SEN)得分和填充质量得分(IQS)等评估指标而言,我们改进的AE填充模型取得了与现有SCDA模型相当或更好的性能。以HLA数据的填充结果为例,AE模型在缺失率为10%和20%时,平均CR分别为0.9468和0.9459,海林格得分分别为0.9765和0.9518,SEN得分分别为0.9977和0.9953,IQS分别为0.9515和0.9044。至于LOS数据的结果,在缺失率为20%时,其平均CR为0.9005,海林格得分0.9384,SEN得分0.9940,IQS为0.8681。总之,我们提出的基因型填充方法在提高GWAS的统计效能和改进GWAS下游分析方面具有巨大潜力。