Feng Zhenxing, Hu Xiuzhen
Department of Sciences, Inner Mongolia University of Technology, Hohhot, China.
Biomed Res Int. 2014;2014:262850. doi: 10.1155/2014/262850. Epub 2014 Jul 21.
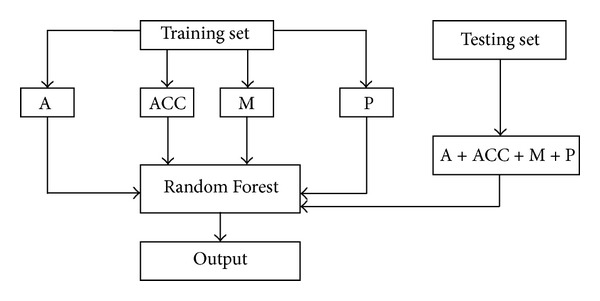
The recognition of protein folds is an important step for the prediction of protein structure and function. After the recognition of 27-class protein folds in 2001 by Ding and Dubchak, prediction algorithms, prediction parameters, and new datasets for the prediction of protein folds have been improved. However, the influences of interactions from predicted secondary structure segments and motif information on protein folding have not been considered. Therefore, the recognition of 27-class protein folds with the interaction of segments and motif information is very important. Based on the 27-class folds dataset built by Liu et al., amino acid composition, the interactions of secondary structure segments, motif frequency, and predicted secondary structure information were extracted. Using the Random Forest algorithm and the ensemble classification strategy, 27-class protein folds and corresponding structural classification were identified by independent test. The overall accuracy of the testing set and structural classification measured up to 78.38% and 92.55%, respectively. When the training set and testing set were combined, the overall accuracy by 5-fold cross validation was 81.16%. In order to compare with the results of previous researchers, the method above was tested on Ding and Dubchak's dataset which has been widely used by many previous researchers, and an improved overall accuracy 70.24% was obtained.
蛋白质折叠的识别是预测蛋白质结构和功能的重要一步。2001年丁和杜布恰克识别出27类蛋白质折叠后,用于蛋白质折叠预测的算法、预测参数和新数据集都得到了改进。然而,预测的二级结构片段之间的相互作用以及基序信息对蛋白质折叠的影响尚未得到考虑。因此,考虑片段相互作用和基序信息来识别27类蛋白质折叠非常重要。基于刘等人构建的27类折叠数据集,提取了氨基酸组成、二级结构片段的相互作用、基序频率和预测的二级结构信息。使用随机森林算法和集成分类策略,通过独立测试识别出27类蛋白质折叠及其相应的结构分类。测试集的总体准确率和结构分类分别达到78.38%和92.55%。当训练集和测试集合并时,5折交叉验证的总体准确率为81.16%。为了与之前研究人员的结果进行比较,在丁和杜布恰克的数据集上测试了上述方法,该数据集已被许多之前的研究人员广泛使用,并获得了70.24%的改进总体准确率。