Yan Zhi, Luke Brian T, Tsang Shirley X, Xing Rui, Pan Yuanming, Liu Yixuan, Wang Jinlian, Geng Tao, Li Jiangeng, Lu Youyong
Laboratory of Molecular Oncology, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Peking University Cancer Hospital and Institute, Beijing, People's Republic of China.
Advanced Biomedical Computing Center, Frederick National Laboratory for Cancer Research, Frederick, MD, USA.
Biomark Insights. 2014 Aug 14;9:67-76. doi: 10.4137/BMI.S13059. eCollection 2014.
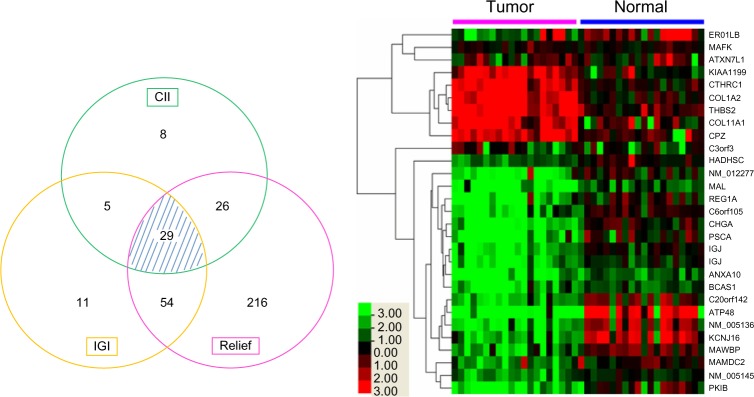
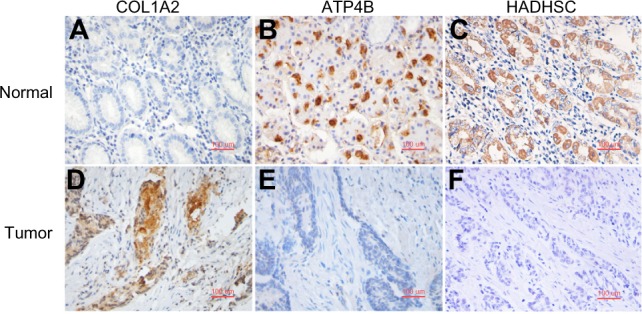
High-throughput gene expression microarrays can be examined by machine-learning algorithms to identify gene signatures that recognize the biological characteristics of specific human diseases, including cancer, with high sensitivity and specificity. A previous study compared 20 gastric cancer (GC) samples against 20 normal tissue (NT) samples and identified 1,519 differentially expressed genes (DEGs). In this study, Classification Information Index (CII), Information Gain Index (IGI), and RELIEF algorithms are used to mine the previously reported gene expression profiling data. In all, 29 of these genes are identified by all three algorithms and are treated as GC candidate biomarkers. Three biomarkers, COL1A2, ATP4B, and HADHSC, are selected and further examined using quantitative real-time polymerase chain reaction (qRT-PCR) and immunohistochemistry (IHC) staining in two independent sets of GC and normal adjacent tissue (NAT) samples. Our study shows that COL1A2 and HADHSC are the two best biomarkers from the microarray data, distinguishing all GC from the NT, whereas ATP4B is diagnostically significant in lab tests because of its wider range of fold-changes in expression. Herein, a data-mining model applicable for small sample sizes is presented and discussed. Our result suggested that this mining model may be useful in small sample-size studies to identify putative biomarkers and potential biological features of GC.
高通量基因表达微阵列可通过机器学习算法进行检测,以识别能够以高灵敏度和特异性识别包括癌症在内的特定人类疾病生物学特征的基因特征。先前的一项研究将20个胃癌(GC)样本与20个正常组织(NT)样本进行比较,鉴定出1519个差异表达基因(DEG)。在本研究中,使用分类信息指数(CII)、信息增益指数(IGI)和RELIEF算法挖掘先前报道的基因表达谱数据。总共,这三种算法共同鉴定出29个基因,并将其视为GC候选生物标志物。选择三种生物标志物COL1A2、ATP4B和HADHSC,并在两组独立的GC和正常相邻组织(NAT)样本中使用定量实时聚合酶链反应(qRT-PCR)和免疫组织化学(IHC)染色进行进一步检测。我们的研究表明,COL1A2和HADHSC是微阵列数据中最好的两种生物标志物,可将所有GC与NT区分开来,而ATP4B因其表达变化倍数范围更广,在实验室检测中具有诊断意义。在此,提出并讨论了一种适用于小样本量的数据挖掘模型。我们的结果表明,这种挖掘模型可能有助于小样本量研究中识别GC的假定生物标志物和潜在生物学特征。