Hao Wanjun, Fan Shihang, Hua Wei, Wang Hanzhong
Key Laboratory for Biological Sciences of Oil Crops, Ministry of Agriculture, Oil Crops Research Institute, Chinese Academy of Agriculture Sciences, Wuhan, China.
Key Laboratory for Biological Sciences of Oil Crops, Ministry of Agriculture, Oil Crops Research Institute, Chinese Academy of Agriculture Sciences, Wuhan, China; College of Life Sciences, Hubei University, Wuhan, China.
PLoS One. 2014 Sep 24;9(9):e108291. doi: 10.1371/journal.pone.0108291. eCollection 2014.
In conventional approaches to plastid and mitochondrial genome sequencing, the sequencing steps are performed separately; thus, plastid DNA (ptDNA) and mitochondrial DNA (mtDNA) should be prepared independently. However, it is difficult to extract pure ptDNA and mtDNA from plant tissue. Following the development of high-throughput sequencing technology, many researchers have attempted to obtain plastid genomes or mitochondrial genomes using high-throughput sequencing data from total DNA. Unfortunately, the huge datasets generated consume massive computing and storage resources and cost a great deal, and even more importantly, excessive pollution reads affect the accuracy of the assembly. Therefore, it is necessary to develop an effective method that can generate base sequences from plant tissue and that is suitable for all plant species. Here, we describe a highly effective, low-cost method for obtaining plastid and mitochondrial genomes simultaneously.
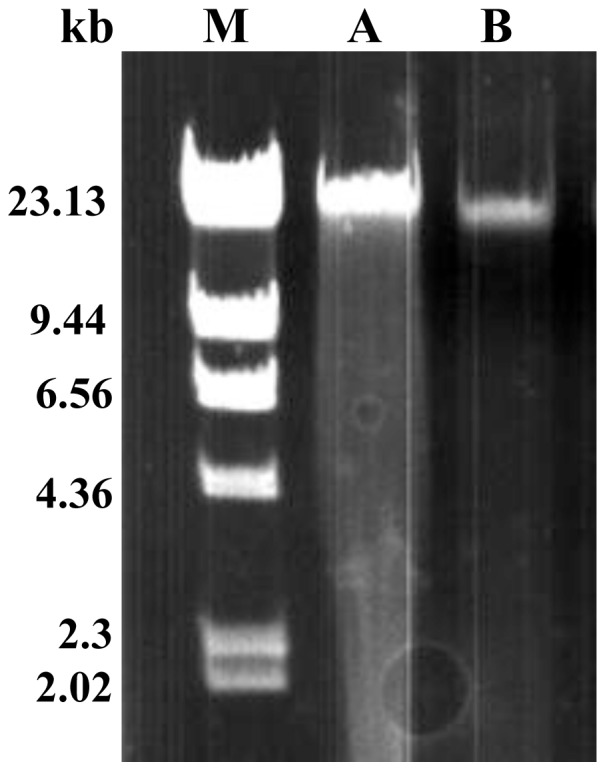
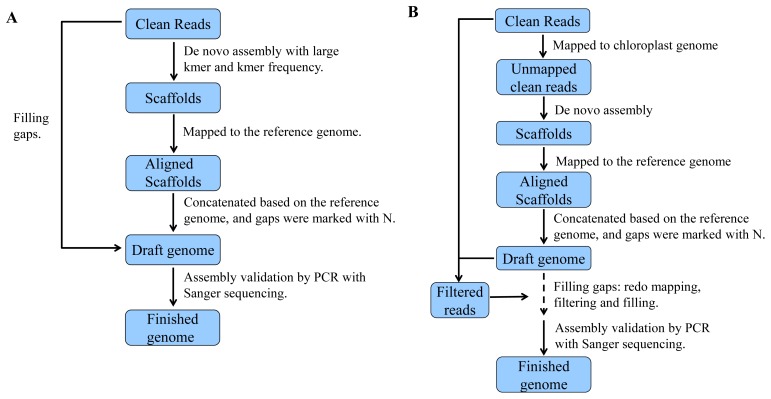
First, we obtained high-quality DNA employing Partial Concentration Extraction. Second, we evaluated the purity of the DNA sample and determined the sequencing dataset size employing Vector Control Quantitative Analysis. Third, paired-end reads were obtained using a high-throughput sequencing platform. Fourth, we obtained scaffolds employing Two-step Assembly. Finally, we filled in gaps using specific methods and obtained complete plastid and mitochondrial genomes. To ensure the accuracy of plastid and mitochondrial genomes, we validated the assembly using PCR and Sanger sequencing. Using this method,we obtained complete plastid and mitochondrial genomes with lengths of 153,533 nt and 223,412 nt separately.
A simple method for extracting, evaluating, sequencing and assembling plastid and mitochondrial genomes was developed. This method has many advantages: it is timesaving, inexpensive and reproducible and produces high-quality sequence. Furthermore, this method can produce plastid and mitochondrial genomes simultaneously and be used for other plant species. Due to its simplicity and extensive applicability, this method will support research on plant cytoplasmic genomes.
在传统的质体和线粒体基因组测序方法中,测序步骤是分开进行的;因此,质体DNA(ptDNA)和线粒体DNA(mtDNA)需要独立制备。然而,从植物组织中提取纯的ptDNA和mtDNA很困难。随着高通量测序技术的发展,许多研究人员尝试利用总DNA的高通量测序数据来获得质体基因组或线粒体基因组。不幸的是,生成的大量数据集消耗了大量的计算和存储资源,成本很高,更重要的是,过多的污染 reads 会影响组装的准确性。因此,有必要开发一种有效的方法,该方法可以从植物组织中生成碱基序列,并且适用于所有植物物种。在这里,我们描述了一种高效、低成本的同时获得质体和线粒体基因组的方法。
首先,我们采用部分浓缩提取法获得了高质量的DNA。其次,我们评估了DNA样品的纯度,并使用载体对照定量分析法确定了测序数据集的大小。第三,使用高通量测序平台获得双端 reads。第四,我们采用两步组装法获得了支架。最后,我们使用特定方法填补缺口,获得了完整的质体和线粒体基因组。为确保质体和线粒体基因组的准确性,我们使用PCR和桑格测序对组装进行了验证。使用这种方法,我们分别获得了长度为153,533 nt和223,412 nt的完整质体和线粒体基因组。
开发了一种提取、评估、测序和组装质体和线粒体基因组的简单方法。该方法具有许多优点:省时、廉价、可重复且能产生高质量序列。此外,该方法可以同时产生质体和线粒体基因组,并可用于其他植物物种。由于其简单性和广泛的适用性,该方法将支持对植物细胞质基因组的研究。