BMC Bioinformatics. 2014;15 Suppl 9(Suppl 9):S2. doi: 10.1186/1471-2105-15-S9-S2. Epub 2014 Sep 10.
There is a widening gap between the throughput of massive parallel sequencing machines and the ability to analyze these sequencing data. Traditional assembly methods requiring long execution time and large amount of memory on a single workstation limit their use on these massive data.
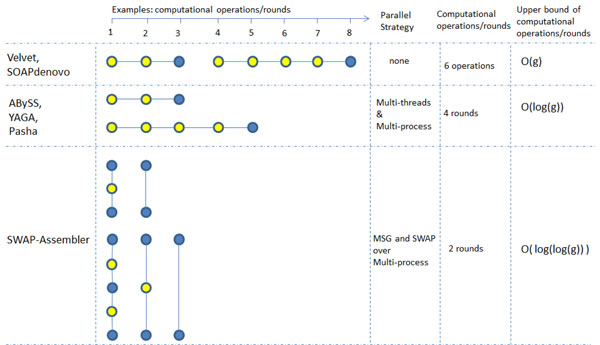
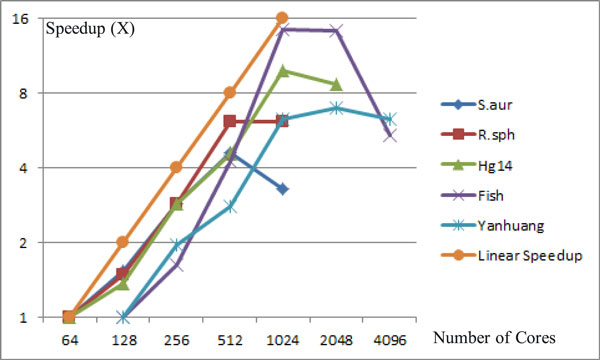
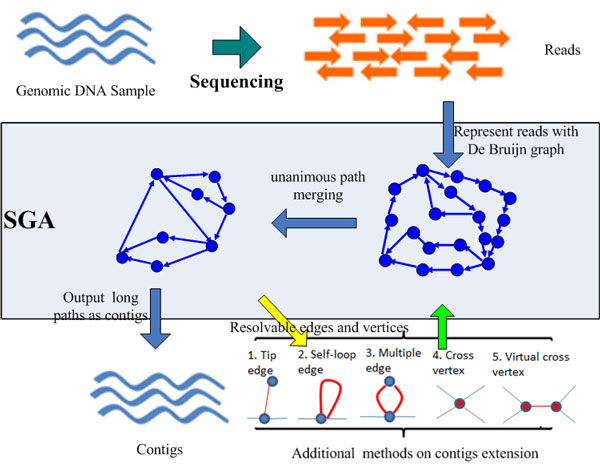
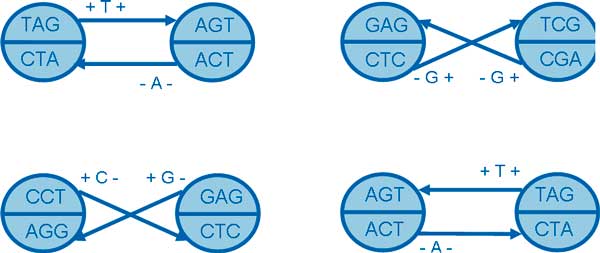
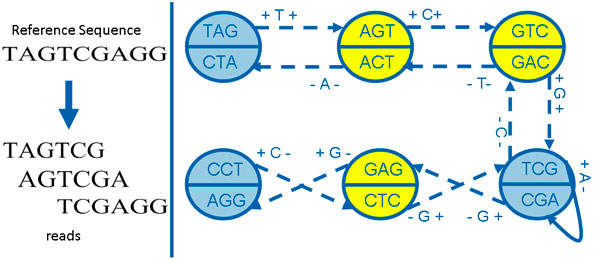
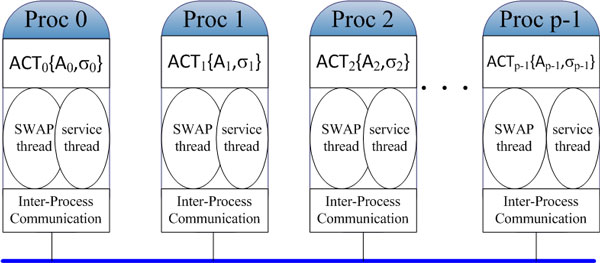
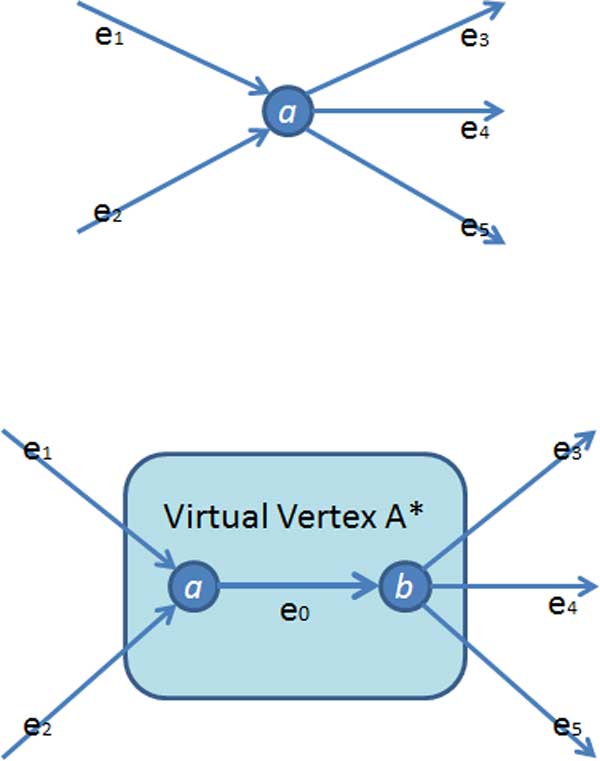
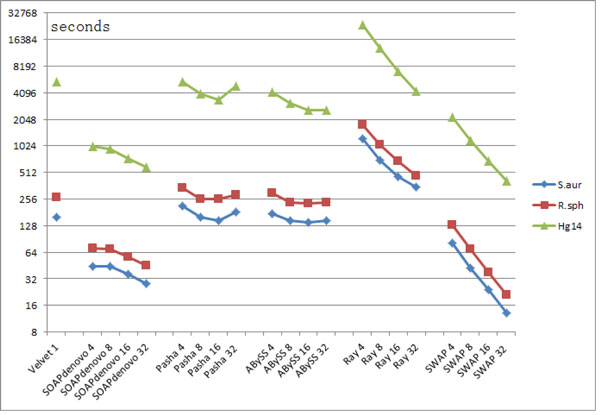
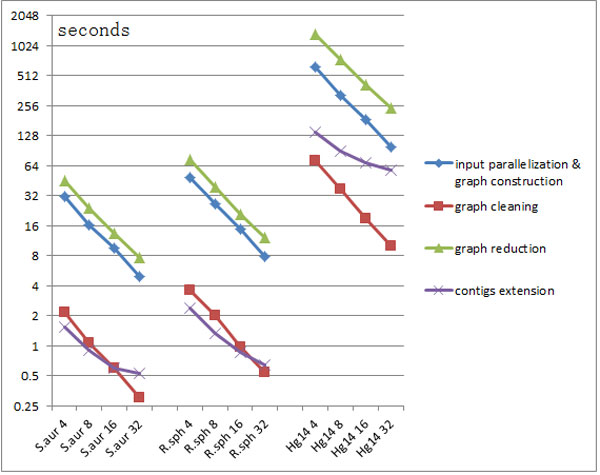
This paper presents a highly scalable assembler named as SWAP-Assembler for processing massive sequencing data using thousands of cores, where SWAP is an acronym for Small World Asynchronous Parallel model. In the paper, a mathematical description of multi-step bi-directed graph (MSG) is provided to resolve the computational interdependence on merging edges, and a highly scalable computational framework for SWAP is developed to automatically preform the parallel computation of all operations. Graph cleaning and contig extension are also included for generating contigs with high quality. Experimental results show that SWAP-Assembler scales up to 2048 cores on Yanhuang dataset using only 26 minutes, which is better than several other parallel assemblers, such as ABySS, Ray, and PASHA. Results also show that SWAP-Assembler can generate high quality contigs with good N50 size and low error rate, especially it generated the longest N50 contig sizes for Fish and Yanhuang datasets.
In this paper, we presented a highly scalable and efficient genome assembly software, SWAP-Assembler. Compared with several other assemblers, it showed very good performance in terms of scalability and contig quality. This software is available at: https://sourceforge.net/projects/swapassembler.
高通量并行测序仪的产出与分析这些测序数据的能力之间存在着越来越大的差距。传统的组装方法需要在单个工作站上执行很长的时间和消耗大量的内存,这限制了它们在这些大规模数据上的使用。
本文提出了一种名为 SWAP-Assembler 的高度可扩展的组装器,用于使用数千个内核处理大规模测序数据,其中 SWAP 是小世界异步并行模型的缩写。本文提供了多步双向图 (MSG) 的数学描述,以解决在合并边时的计算相关性,并开发了一种高度可扩展的 SWAP 计算框架,用于自动执行所有操作的并行计算。还包括图形清理和重叠群扩展,以生成高质量的重叠群。实验结果表明,SWAP-Assembler 在使用 26 分钟的情况下,在 Yanhuang 数据集上可扩展到 2048 个内核,优于其他一些并行组装器,如 ABySS、Ray 和 PASHA。结果还表明,SWAP-Assembler 可以生成高质量的重叠群,具有良好的 N50 大小和低错误率,特别是它为 Fish 和 Yanhuang 数据集生成了最长的 N50 重叠群大小。
本文提出了一种高度可扩展和高效的基因组组装软件 SWAP-Assembler。与其他一些组装器相比,它在可扩展性和重叠群质量方面表现出了非常好的性能。该软件可在以下网址获得:https://sourceforge.net/projects/swapassembler。