Genome Center, UC, Davis, CA 95616, USA.
Gigascience. 2013 Jul 22;2(1):10. doi: 10.1186/2047-217X-2-10.
The process of generating raw genome sequence data continues to become cheaper, faster, and more accurate. However, assembly of such data into high-quality, finished genome sequences remains challenging. Many genome assembly tools are available, but they differ greatly in terms of their performance (speed, scalability, hardware requirements, acceptance of newer read technologies) and in their final output (composition of assembled sequence). More importantly, it remains largely unclear how to best assess the quality of assembled genome sequences. The Assemblathon competitions are intended to assess current state-of-the-art methods in genome assembly.
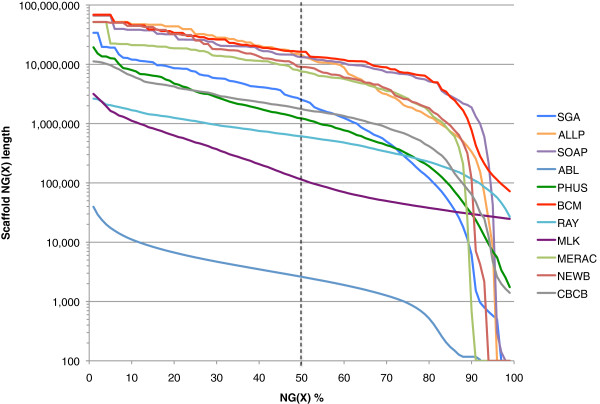
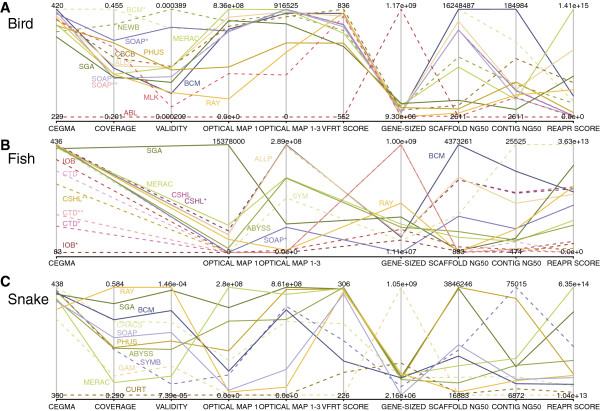
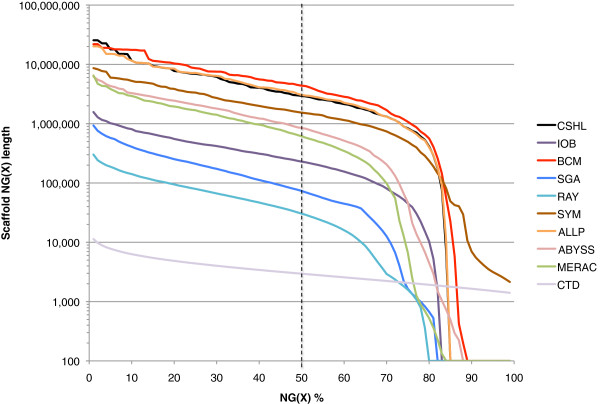
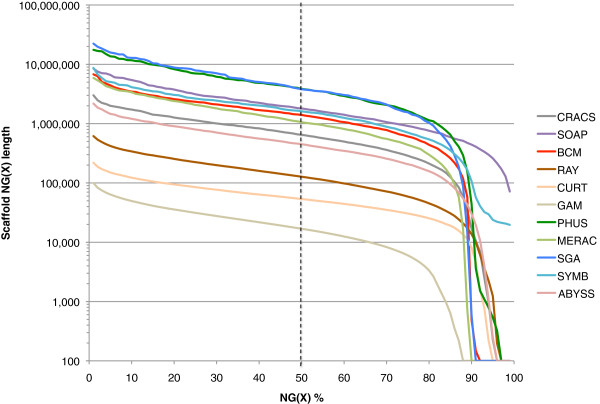
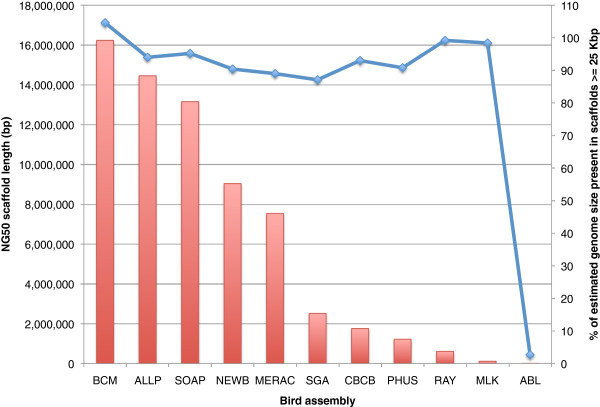
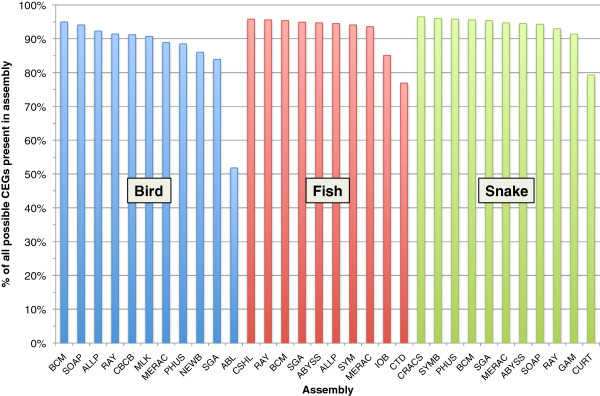
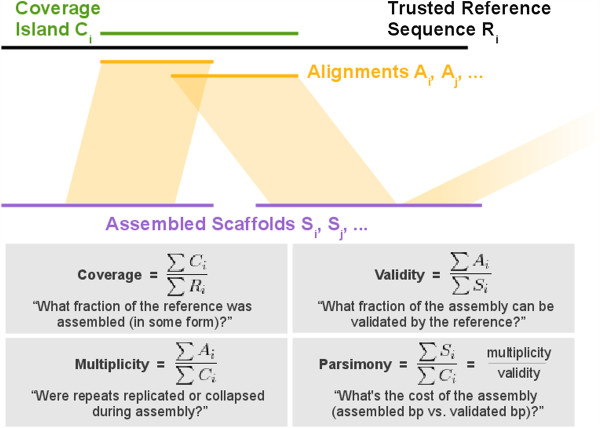
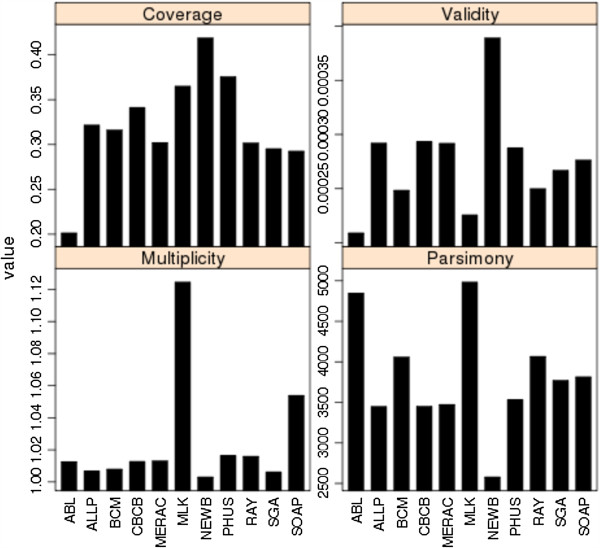
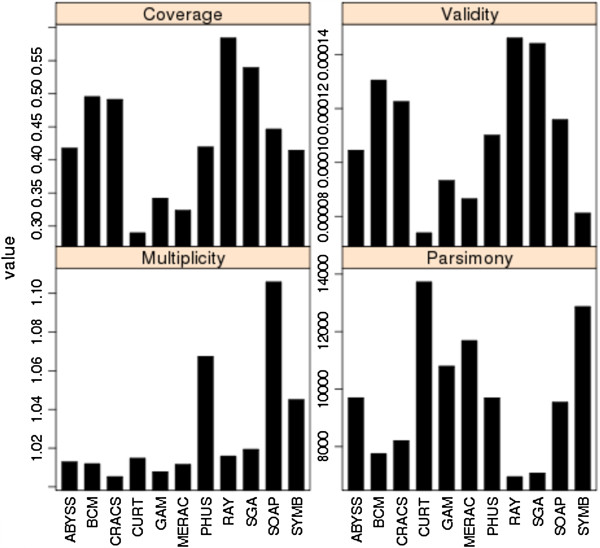
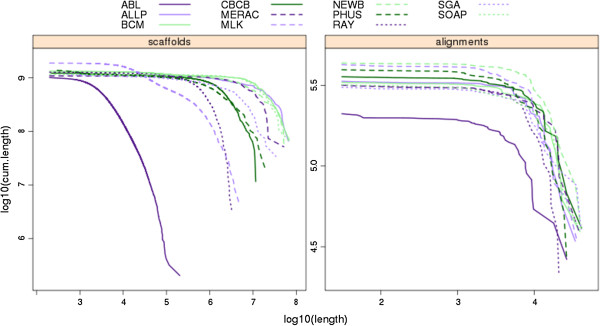
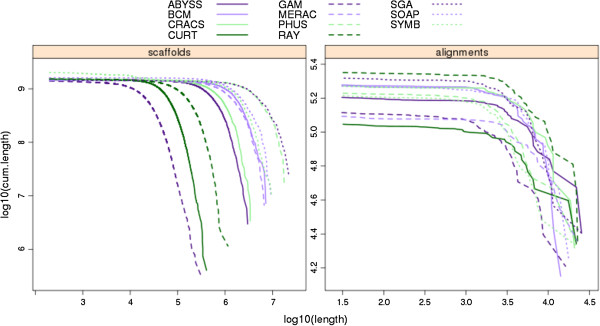
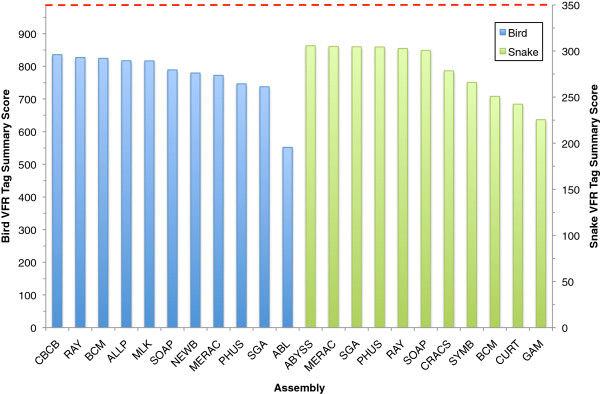
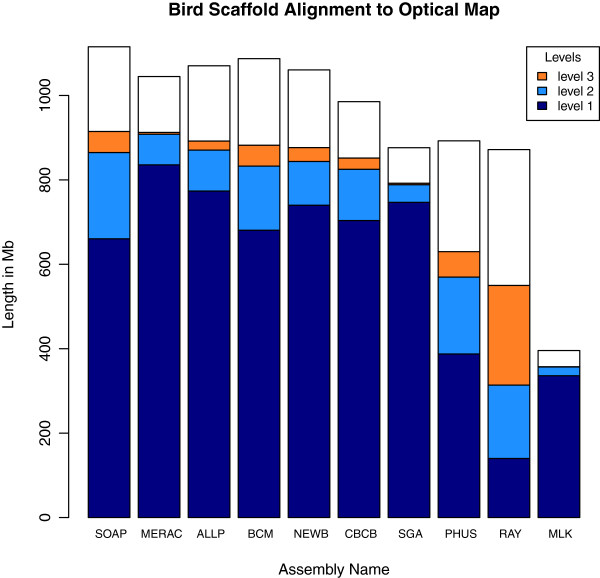
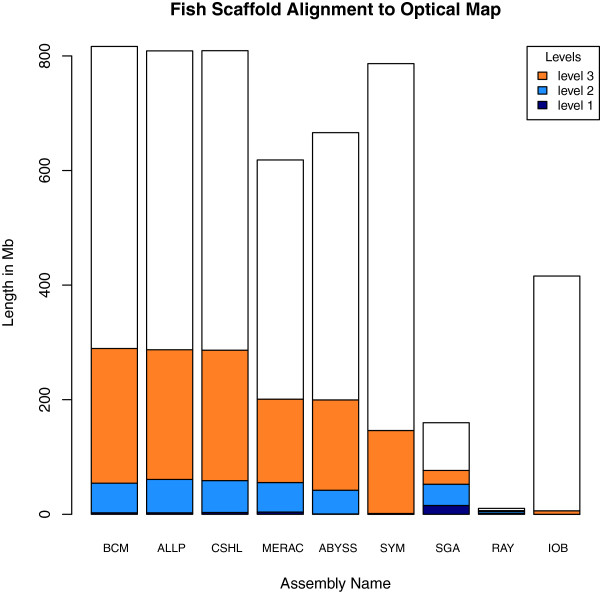
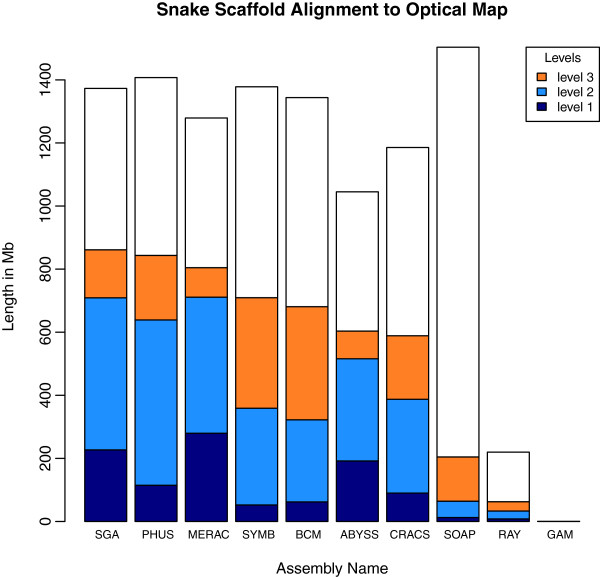
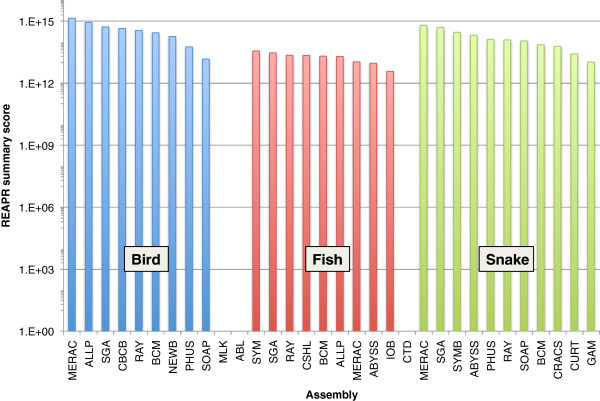
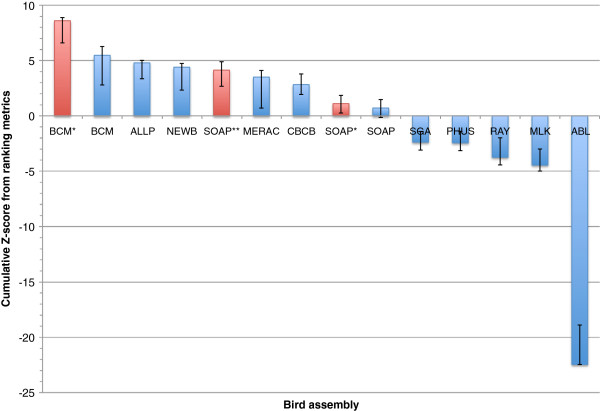
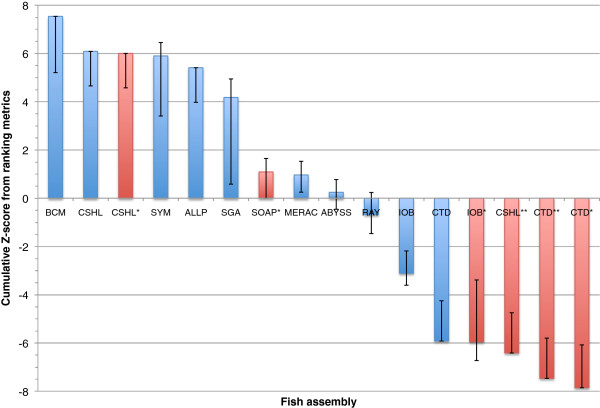
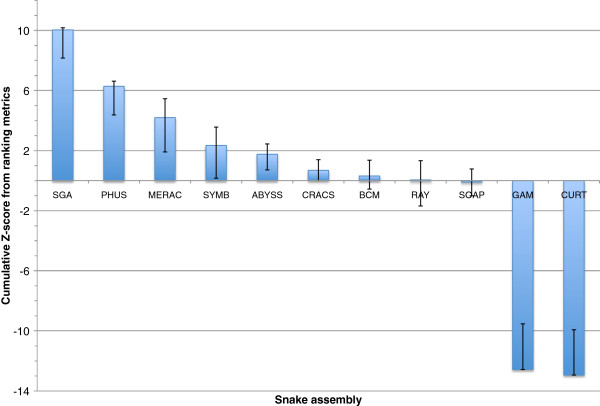
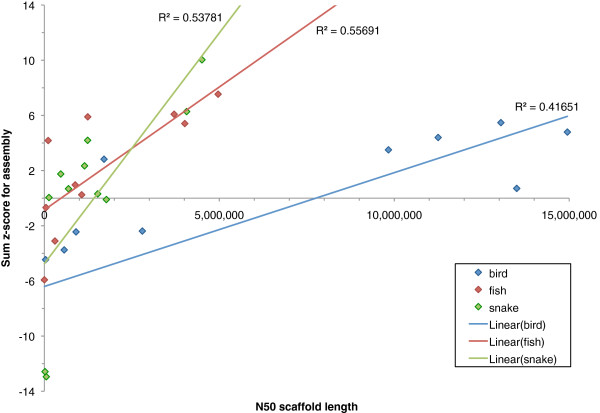
In Assemblathon 2, we provided a variety of sequence data to be assembled for three vertebrate species (a bird, a fish, and snake). This resulted in a total of 43 submitted assemblies from 21 participating teams. We evaluated these assemblies using a combination of optical map data, Fosmid sequences, and several statistical methods. From over 100 different metrics, we chose ten key measures by which to assess the overall quality of the assemblies.
Many current genome assemblers produced useful assemblies, containing a significant representation of their genes and overall genome structure. However, the high degree of variability between the entries suggests that there is still much room for improvement in the field of genome assembly and that approaches which work well in assembling the genome of one species may not necessarily work well for another.
生成原始基因组序列数据的过程继续变得更便宜、更快、更准确。然而,将这些数据组装成高质量的完成基因组序列仍然具有挑战性。有许多基因组组装工具可用,但它们在性能(速度、可扩展性、硬件要求、对较新技术的接受程度)和最终输出(组装序列的组成)方面有很大的不同。更重要的是,如何最好地评估组装基因组序列的质量在很大程度上仍不清楚。Assemblathon 竞赛旨在评估基因组组装的当前最先进方法。
在 Assemblathon 2 中,我们提供了多种序列数据供三种脊椎动物物种(鸟类、鱼类和蛇类)进行组装。这总共产生了来自 21 个参赛团队的 43 个提交的组装结果。我们使用光学图谱数据、Fosmid 序列和几种统计方法来评估这些组装结果。在 100 多种不同的指标中,我们选择了十个关键指标来评估组装的整体质量。
许多当前的基因组组装器生成了有用的组装结果,其中包含了其基因和整体基因组结构的重要代表。然而,这些条目之间高度的可变性表明,在基因组组装领域仍有很大的改进空间,并且在组装一种物种的基因组方面表现良好的方法不一定在另一种物种上也能很好地发挥作用。