Li Qianqian, Liu Jianguo, Zhang Litao, Liu Qian
Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China; University of the Chinese Academy of Sciences, Beijing, China.
Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China.
PLoS One. 2014 Sep 25;9(9):e108488. doi: 10.1371/journal.pone.0108488. eCollection 2014.
Algae in the order Trentepohliales have a broad geographic distribution and are generally characterized by the presence of abundant β-carotene. The many monographs published to date have mainly focused on their morphology, taxonomy, phylogeny, distribution and reproduction; molecular studies of this order are still rare. High-throughput RNA sequencing (RNA-Seq) technology provides a powerful and efficient method for transcript analysis and gene discovery in Trentepohlia jolithus.
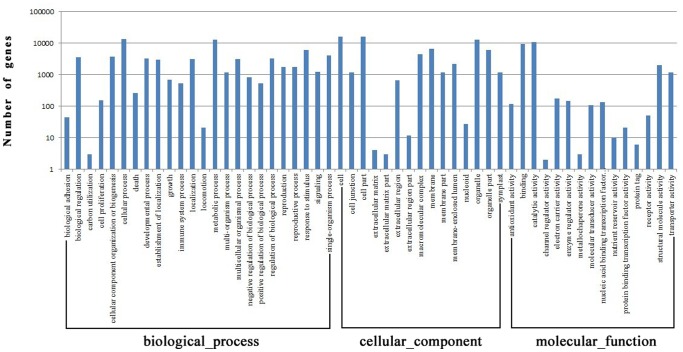
METHODS/PRINCIPAL FINDINGS: Illumina HiSeq 2000 sequencing generated 55,007,830 Illumina PE raw reads, which were assembled into 41,328 assembled unigenes. Based on NR annotation, 53.28% of the unigenes (22,018) could be assigned to gene ontology classes with 54 subcategories and 161,451 functional terms. A total of 26,217 (63.44%) assembled unigenes were mapped to 128 KEGG pathways. Furthermore, a set of 5,798 SSRs in 5,206 unigenes and 131,478 putative SNPs were identified. Moreover, the fact that all of the C4 photosynthesis genes exist in T. jolithus suggests a complex carbon acquisition and fixation system. Similarities and differences between T. jolithus and other algae in carotenoid biosynthesis are also described in depth.
CONCLUSIONS/SIGNIFICANCE: This is the first broad transcriptome survey for T. jolithus, increasing the amount of molecular data available for the class Ulvophyceae. As well as providing resources for functional genomics studies, the functional genes and putative pathways identified here will contribute to a better understanding of carbon fixation and fatty acid and carotenoid biosynthesis in T. jolithus.
橘色集球藻目藻类具有广泛的地理分布,其一般特征是含有丰富的β-胡萝卜素。迄今为止出版的众多专著主要聚焦于它们的形态、分类、系统发育、分布和繁殖;对该目的分子研究仍然很少。高通量RNA测序(RNA-Seq)技术为橘色集球藻的转录本分析和基因发现提供了一种强大而高效的方法。
方法/主要发现:Illumina HiSeq 2000测序产生了55,007,830条Illumina PE原始读段,这些读段被组装成41,328个组装单基因。基于NR注释,53.28%的单基因(22,018个)可被归入具有54个子类别和161,451个功能术语的基因本体类别。总共26,217个(63.44%)组装单基因被映射到128条KEGG通路。此外,在5,206个单基因中鉴定出了一组5,798个SSR和131,478个推定的SNP。而且,所有C4光合作用基因都存在于橘色集球藻中这一事实表明其具有复杂的碳获取和固定系统。还深入描述了橘色集球藻与其他藻类在类胡萝卜素生物合成方面的异同。
结论/意义:这是对橘色集球藻的首次广泛转录组调查,增加了绿藻纲可用的分子数据量。除了为功能基因组学研究提供资源外,这里鉴定出的功能基因和推定通路将有助于更好地理解橘色集球藻中的碳固定以及脂肪酸和类胡萝卜素生物合成。