Nguyen Hai, Maier James, Huang He, Perrone Victoria, Simmerling Carlos
Department of Chemistry, ‡Laufer Center for Physical and Quantitative Biology and §Graduate Program in Biochemistry and Structural Biology, Stony Brook University , Stony Brook, New York 11794-5252, United States.
J Am Chem Soc. 2014 Oct 8;136(40):13959-62. doi: 10.1021/ja5032776. Epub 2014 Sep 25.
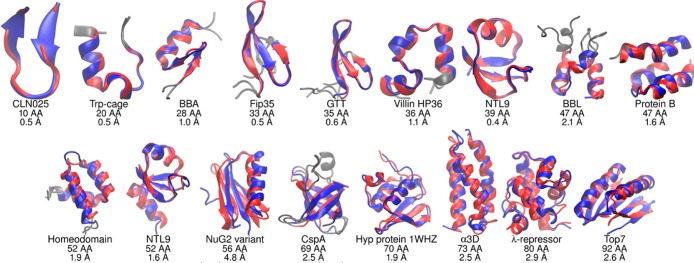
The millisecond time scale needed for molecular dynamics simulations to approach the quantitative study of protein folding is not yet routine. One approach to extend the simulation time scale is to perform long simulations on specialized and expensive supercomputers such as Anton. Ideally, however, folding simulations would be more economical while retaining reasonable accuracy, and provide feedback on structure, stability and function rapidly enough if partnered directly with experiment. Approaches to this problem typically involve varied compromises between accuracy, precision, and cost; the goal here is to address whether simple implicit solvent models have become sufficiently accurate for their weaknesses to be offset by their ability to rapidly provide much more precise conformational data as compared to explicit solvent. We demonstrate that our recently developed physics-based model performs well on this challenge, enabling accurate all-atom simulated folding for 16 of 17 proteins with a variety of sizes, secondary structure, and topologies. The simulations were carried out using the Amber software on inexpensive GPUs, providing ∼1 μs/day per GPU, and >2.5 ms data presented here. We also show that native conformations are preferred over misfolded structures for 14 of the 17 proteins. For the other 3, misfolded structures are thermodynamically preferred, suggesting opportunities for further improvement.
分子动力学模拟达到蛋白质折叠定量研究所需的毫秒时间尺度尚未成为常规操作。扩展模拟时间尺度的一种方法是在诸如Anton等专门且昂贵的超级计算机上进行长时间模拟。然而,理想情况下,折叠模拟应更经济,同时保持合理的准确性,并在与实验直接结合时能足够快速地提供有关结构、稳定性和功能的反馈。解决此问题的方法通常涉及在准确性、精确性和成本之间进行各种权衡;这里的目标是探讨简单的隐式溶剂模型是否已足够准确,以至于其弱点能被其与显式溶剂相比快速提供更精确构象数据的能力所抵消。我们证明,我们最近开发的基于物理的模型在这一挑战中表现良好,能够对17种具有各种大小、二级结构和拓扑结构的蛋白质中的16种进行准确的全原子模拟折叠。模拟使用Amber软件在廉价的图形处理器(GPU)上进行,每个GPU每天可提供约1微秒的模拟时间,此处展示的数据超过2.5毫秒。我们还表明,17种蛋白质中的14种,其天然构象比错误折叠的结构更受青睐。对于另外3种蛋白质,错误折叠的结构在热力学上更受青睐,这表明仍有进一步改进的空间。