Ghirardello Stefano, Dusi Elisa, Castiglione Bianca, Fumagalli Monica, Mosca Fabio
Neonatal Intensive Care Unit Department of Clinical Sciences and Community Health, University of Milan, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Ital J Pediatr. 2014 Sep 26;40:76. doi: 10.1186/s13052-014-0076-4.
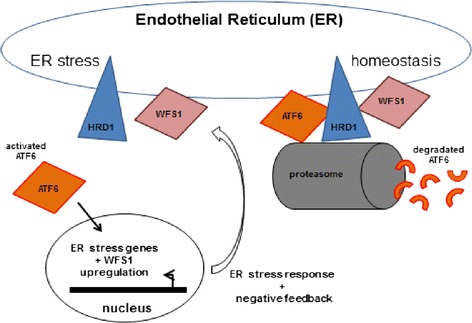
Wolfram syndrome (WS) is an autosomal recessive neurodegenerative disorder characterized by diabetes mellitus (DM), optic atrophy (OA), central diabetes insipidus (CDI) and deafness (D). The phenotype of the disease has been associated with several mutations in the WFS1 gene, a nuclear gene localized on chromosome 4. Since the discovery of the association between WFS1 gene and Wolfram syndrome, more than 150 mutations have been identified in WS patients. We previously described the first case of perinatal onset of Wolfram syndrome newborn carrying a segmental uniparental heterodysomy affecting the short arm of chromosome 4 responsible for a significant reduction in wolframin expression. Here we review and discuss the pathophysiological mechanisms that we believe responsible for the perinatal onset of Wolfram syndrome as these data strongly suggest a role for WFS1 gene in foetal and neonatal neurodevelopment.
We described a male patient of 30 weeks' gestation with intrauterine growth restriction and poly-hydramnios. During the first days of life, the patient showed a 19% weight loss associated with polyuria and hypernatremia. The presence of persistent hypernatremia (serum sodium 150 mEq/L), high plasma osmolarity (322 mOsm/L) and low urine osmolarity (190 mOsm/l) with a Uosm/Posm ratio < 1 were consistent with CDI. The diagnosis of CDI was confirmed by the desmopressin test and the brain magnetic resonance imaging (MRI) at 34 weeks of age, that showed the lack of posterior pituitary hyperintense signal. In addition, a bilateral asymmetrical optic nerve hypoplasia associated with right orbital bone hypoplasia was observed, suggesting the diagnosis of WF. During the five years follow-up the patient did not developed glucose intolerance or diabetes mellitus. By the end of the second year of life, primary non-autoimmune central hypothyroidism and mild neurodevelopment retardation were diagnosed.
The analysis of our case, in the light of the most recent literature, suggests a possible role for WFS1 gene in the development of certain brain structures during the fetal period. Wolfram syndrome should be considered in the differential diagnosis of the rare cases of congenital central diabetes insipidus developed in the neonatal period.
沃夫勒姆综合征(WS)是一种常染色体隐性神经退行性疾病,其特征为糖尿病(DM)、视神经萎缩(OA)、中枢性尿崩症(CDI)和耳聋(D)。该疾病的表型与WFS1基因的多种突变有关,WFS1基因是一种位于4号染色体上的核基因。自发现WFS1基因与沃夫勒姆综合征之间的关联以来,在WS患者中已鉴定出150多种突变。我们之前描述了首例围产期发病的沃夫勒姆综合征新生儿,其携带影响4号染色体短臂的节段性单亲异源二体性,导致沃尔弗胺表达显著降低。在此,我们回顾并讨论我们认为导致沃夫勒姆综合征围产期发病的病理生理机制,因为这些数据强烈表明WFS1基因在胎儿和新生儿神经发育中起作用。
我们描述了一名妊娠30周的男性患者,伴有宫内生长受限和羊水过多。在出生后的头几天,该患者体重下降了19%,伴有多尿和高钠血症。持续高钠血症(血清钠150 mEq/L)、高血浆渗透压(322 mOsm/L)和低尿渗透压(190 mOsm/l)且尿渗/血渗比值<1,与中枢性尿崩症一致。去氨加压素试验和34周龄时的脑磁共振成像(MRI)证实了中枢性尿崩症的诊断,该检查显示垂体后叶缺乏高强度信号。此外,观察到双侧不对称性视神经发育不全并伴有右眼眶骨发育不全,提示沃夫勒姆综合征的诊断。在五年的随访中,该患者未出现葡萄糖不耐受或糖尿病。到生命的第二年年底,诊断出原发性非自身免疫性中枢性甲状腺功能减退和轻度神经发育迟缓。
根据最新文献对我们病例的分析表明,WFS1基因在胎儿期某些脑结构的发育中可能起作用。在新生儿期发生的先天性中枢性尿崩症的罕见病例的鉴别诊断中,应考虑沃夫勒姆综合征。