Matsunaga Kimie, Tanabe Katsuya, Inoue Hiroshi, Okuya Shigeru, Ohta Yasuharu, Akiyama Masaru, Taguchi Akihiko, Kora Yukari, Okayama Naoko, Yamada Yuichiro, Wada Yasuhiko, Amemiya Shin, Sugihara Shigetaka, Nakao Yuzo, Oka Yoshitomo, Tanizawa Yukio
Division of Endocrinology, Metabolism, Hematological Science and Therapeutics, Yamaguchi University Graduate School of Medicine, Ube, Yamaguchi, Japan.
Division of Laboratory Yamaguchi University Hospital, Ube, Yamaguchi, Japan.
PLoS One. 2014 Sep 11;9(9):e106906. doi: 10.1371/journal.pone.0106906. eCollection 2014.
Wolfram syndrome (WFS) is a recessive neurologic and endocrinologic degenerative disorder, and is also known as DIDMOAD (Diabetes Insipidus, early-onset Diabetes Mellitus, progressive Optic Atrophy and Deafness) syndrome. Most affected individuals carry recessive mutations in the Wolfram syndrome 1 gene (WFS1). However, the phenotypic pleiomorphism, rarity and molecular complexity of this disease complicate our efforts to understand WFS. To address this limitation, we aimed to describe complications and to elucidate the contributions of WFS1 mutations to clinical manifestations in Japanese patients with WFS.
The minimal ascertainment criterion for diagnosing WFS was having both early onset diabetes mellitus and bilateral optic atrophy. Genetic analysis for WFS1 was performed by direct sequencing.
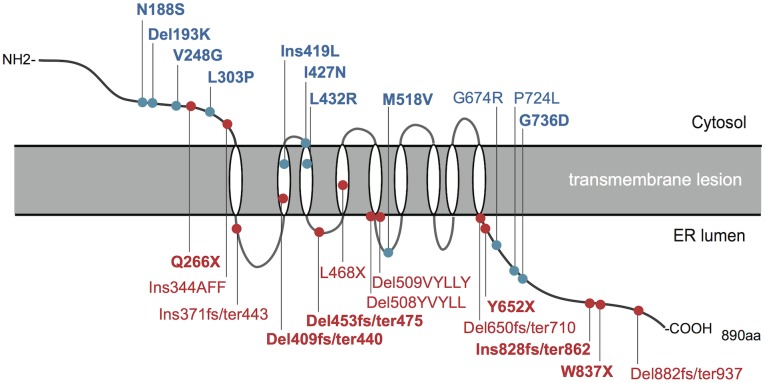
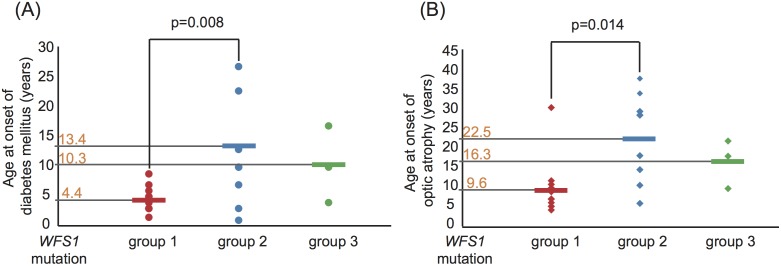
Sixty-seven patients were identified nationally for a prevalence of one per 710,000, with 33 patients (49%) having all 4 components of DIDMOAD. In 40 subjects who agreed to participate in this investigation from 30 unrelated families, the earliest manifestation was DM at a median age of 8.7 years, followed by OA at a median age of 15.8 years. However, either OA or DI was the first diagnosed feature in 6 subjects. In 10, features other than DM predated OA. Twenty-seven patients (67.5%) had a broad spectrum of recessive mutations in WFS1. Two patients had mutations in only one allele. Eleven patients (27.5%) had intact WFS1 alleles. Ages at onset of both DM and OA in patients with recessive WFS1 mutations were indistinguishable from those in patients without WFS1 mutations. In the patients with predicted complete loss-of-function mutations, ages at the onsets of both DM and OA were significantly earlier than those in patients with predicted partial-loss-of function mutations.
CONCLUSION/SIGNIFICANCE: This study emphasizes the clinical and genetic heterogeneity in patients with WFS. Genotype-phenotype correlations may exist in patients with WFS1 mutations, as demonstrated by the disease onset.
沃尔弗拉姆综合征(WFS)是一种隐性神经和内分泌退行性疾病,也被称为 DIDMOAD(尿崩症、早发性糖尿病、进行性视神经萎缩和耳聋)综合征。大多数患者在沃尔弗拉姆综合征 1 基因(WFS1)中携带隐性突变。然而,这种疾病的表型多态性、罕见性和分子复杂性使我们对 WFS 的理解变得复杂。为了解决这一局限性,我们旨在描述日本 WFS 患者的并发症,并阐明 WFS1 突变对临床表现的影响。
诊断 WFS 的最小确诊标准是患有早发性糖尿病和双侧视神经萎缩。通过直接测序对 WFS1 进行基因分析。
全国共确定了 67 例患者,患病率为每 710,000 人中有 1 例,其中 33 例(49%)具有 DIDMOAD 的所有 4 个组成部分。在来自 30 个无关家庭的 40 名同意参与本研究的受试者中,最早的表现是糖尿病,中位年龄为 8.7 岁,其次是视神经萎缩,中位年龄为 15.8 岁。然而,在 6 名受试者中,视神经萎缩或尿崩症是首次诊断出的特征。在 10 名受试者中,糖尿病以外的特征先于视神经萎缩出现。27 例患者(67.5%)在 WFS1 中存在广泛的隐性突变。2 例患者仅一个等位基因发生突变。11 例患者(27.5%)的 WFS1 等位基因完整。隐性 WFS1 突变患者的糖尿病和视神经萎缩发病年龄与无 WFS1 突变患者无差异。在预测为功能完全丧失突变的患者中,糖尿病和视神经萎缩的发病年龄明显早于预测为功能部分丧失突变的患者。
结论/意义:本研究强调了 WFS 患者的临床和遗传异质性。如疾病发病情况所示,WFS1 突变患者可能存在基因型 - 表型相关性。