From the Division of Clinical Pharmacology, Department of Medicine (D.W.T., S.R.T., A.K., M.A.S., H.I., J.W., A.G., M.S.M., W.C., D.G.H.) and Department of Pharmacology and Toxicology, Faculty of Pharmacy (M.A.S.), Mansoura University, Mansoura, Egypt; Departments of Molecular Physiology and Biophysics (A.E.N.) and Biostatistics (C.-I.L., Y.S.), Vanderbilt University School of Medicine, Nashville, TN; and Department of Cell and Molecular Physiology, University of North Carolina, Chapel Hill (W.J.A.).
Hypertension. 2014 Nov;64(5):1108-15. doi: 10.1161/HYPERTENSIONAHA.114.04147. Epub 2014 Aug 4.
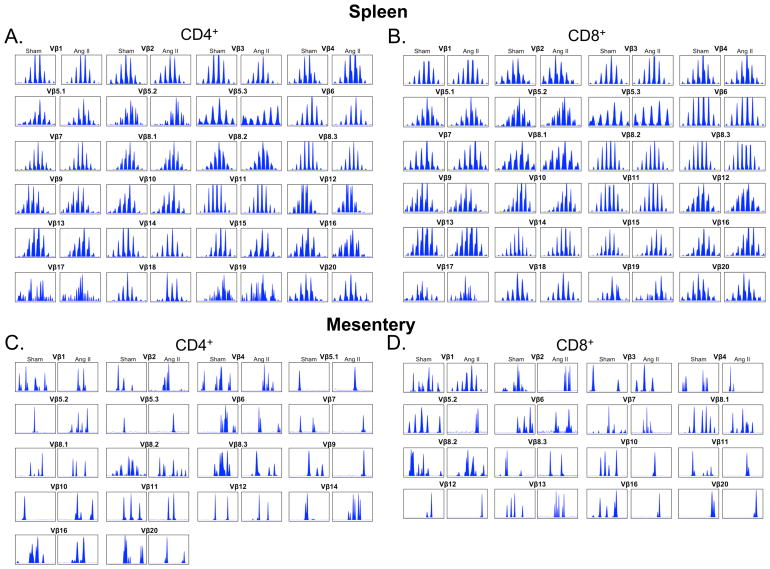
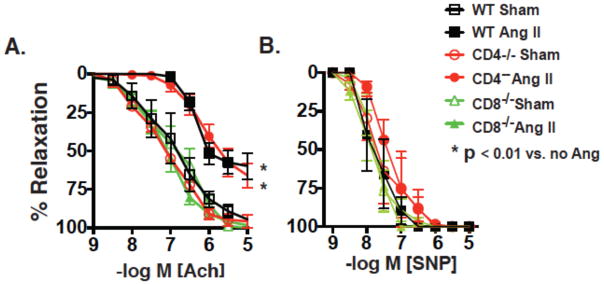
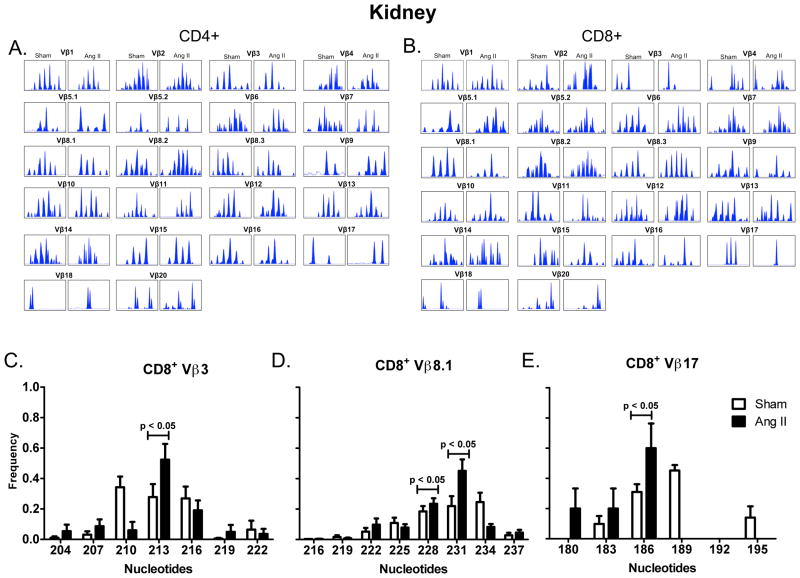
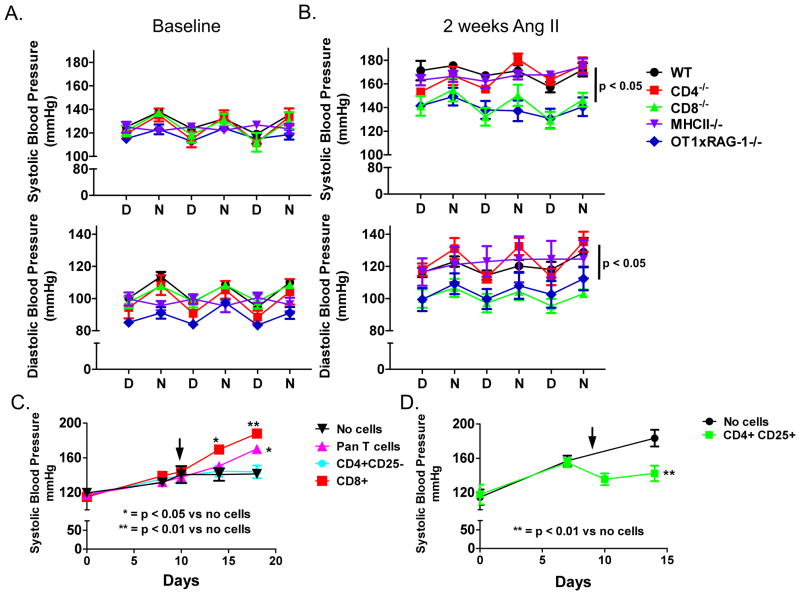
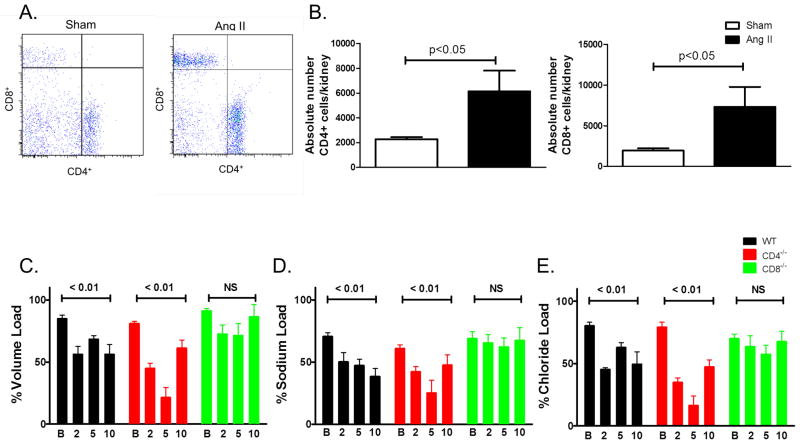
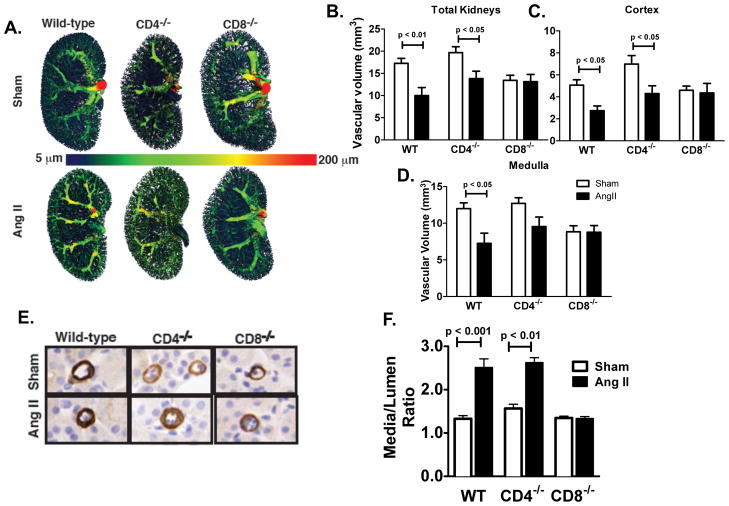
Recent studies have emphasized a role of adaptive immunity, and particularly T cells, in the genesis of hypertension. We sought to determine the T-cell subtypes that contribute to hypertension and renal inflammation in angiotensin II-induced hypertension. Using T-cell receptor spectratyping to examine T-cell receptor usage, we demonstrated that CD8(+) cells, but not CD4(+) cells, in the kidney exhibited altered T-cell receptor transcript lengths in Vβ3, 8.1, and 17 families in response to angiotensin II-induced hypertension. Clonality was not observed in other organs. The hypertension caused by angiotensin II in CD4(-/-) and MHCII(-/-) mice was similar to that observed in wild-type mice, whereas CD8(-/-) mice and OT1xRAG-1(-/-) mice, which have only 1 T-cell receptor, exhibited a blunted hypertensive response to angiotensin II. Adoptive transfer of pan T cells and CD8(+) T cells but not CD4(+)/CD25(-) cells conferred hypertension to RAG-1(-/-) mice. In contrast, transfer of CD4(+)/CD25(+) cells to wild-type mice receiving angiotensin II decreased blood pressure. Mice treated with angiotensin II exhibited increased numbers of kidney CD4(+) and CD8(+) T cells. In response to a sodium/volume challenge, wild-type and CD4(-/-) mice infused with angiotensin II retained water and sodium, whereas CD8(-/-) mice did not. CD8(-/-) mice were also protected against angiotensin-induced endothelial dysfunction and vascular remodeling in the kidney. These data suggest that in the development of hypertension, an oligoclonal population of CD8(+) cells accumulates in the kidney and likely contributes to hypertension by contributing to sodium and volume retention and vascular rarefaction.
最近的研究强调了适应性免疫,特别是 T 细胞,在高血压的发生中的作用。我们试图确定导致血管紧张素 II 诱导的高血压中的高血压和肾脏炎症的 T 细胞亚型。使用 T 细胞受体谱分析来检查 T 细胞受体的使用情况,我们证明了在血管紧张素 II 诱导的高血压中,肾脏中的 CD8(+)细胞而不是 CD4(+)细胞,在 Vβ3、8.1 和 17 家族中表现出改变的 T 细胞受体转录长度。在其他器官中未观察到克隆性。在 CD4(-/-)和 MHCII(-/-)小鼠中,血管紧张素 II 引起的高血压与在野生型小鼠中观察到的高血压相似,而 CD8(-/-)小鼠和 OT1xRAG-1(-/-)小鼠(只有 1 种 T 细胞受体)对血管紧张素 II 的高血压反应减弱。Pan T 细胞和 CD8(+)T 细胞但不是 CD4(+)/CD25(-)细胞的过继转移赋予了 RAG-1(-/-)小鼠高血压。相比之下,将 CD4(+)/CD25(+)细胞转移到接受血管紧张素 II 的野生型小鼠中会降低血压。用血管紧张素 II 处理的小鼠表现出肾脏中 CD4(+)和 CD8(+)T 细胞数量的增加。在对钠/容量挑战的反应中,输注血管紧张素 II 的野生型和 CD4(-/-)小鼠保留水和钠,而 CD8(-/-)小鼠则没有。CD8(-/-)小鼠也免受血管紧张素诱导的内皮功能障碍和肾脏血管重塑的影响。这些数据表明,在高血压的发展过程中,一群寡克隆的 CD8(+)细胞在肾脏中积累,可能通过促进钠和容量保留以及血管稀疏来导致高血压。