From the Division of Clinical Pharmacology (J.W., S.R.T., A.K., D.W.T., M.A.S., L.X., M.S.M., W.C., D.G.H), and Departments of Medicine and Pharmacology (J.W., D.G.H.), Vanderbilt University, Nashville, TN; and Department of Pharmacology and Toxicology, Faculty of Pharmacy, Mansoura University, Egypt (M.A.S.).
Circ Res. 2014 Feb 14;114(4):616-25. doi: 10.1161/CIRCRESAHA.114.302157. Epub 2013 Dec 17.
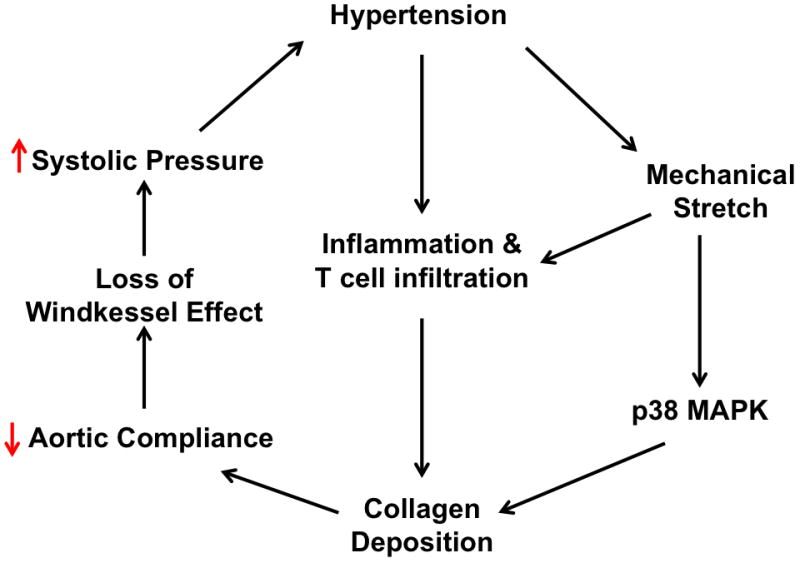
Aortic stiffening commonly occurs in hypertension and further elevates systolic pressure. Hypertension is also associated with vascular inflammation and increased mechanical stretch. The interplay between inflammation, mechanical stretch, and aortic stiffening in hypertension remains undefined.
Our aim was to determine the role of inflammation and mechanical stretch in aortic stiffening.
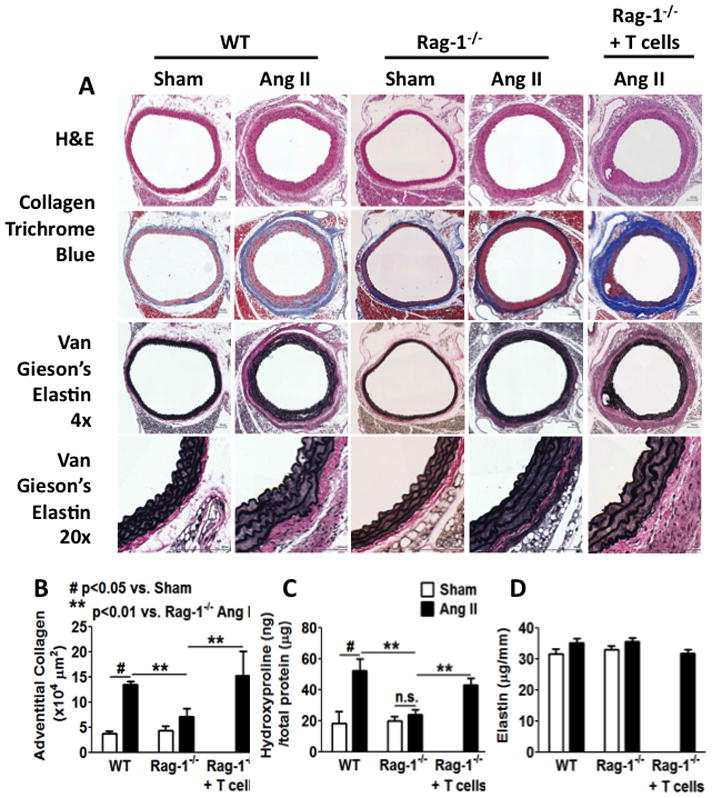
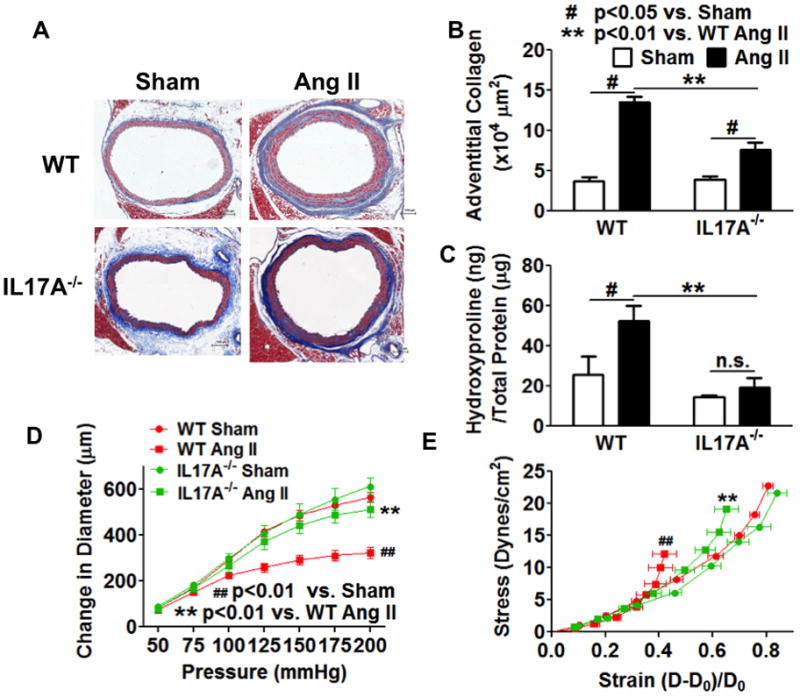
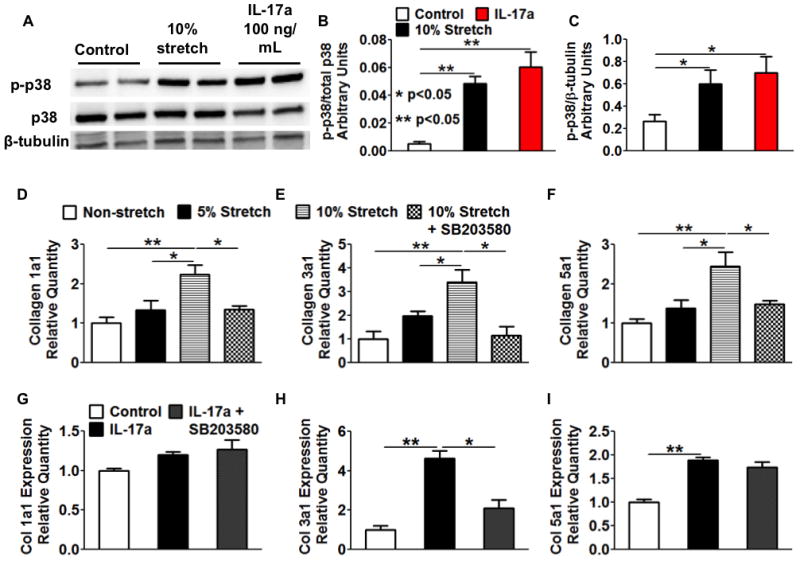
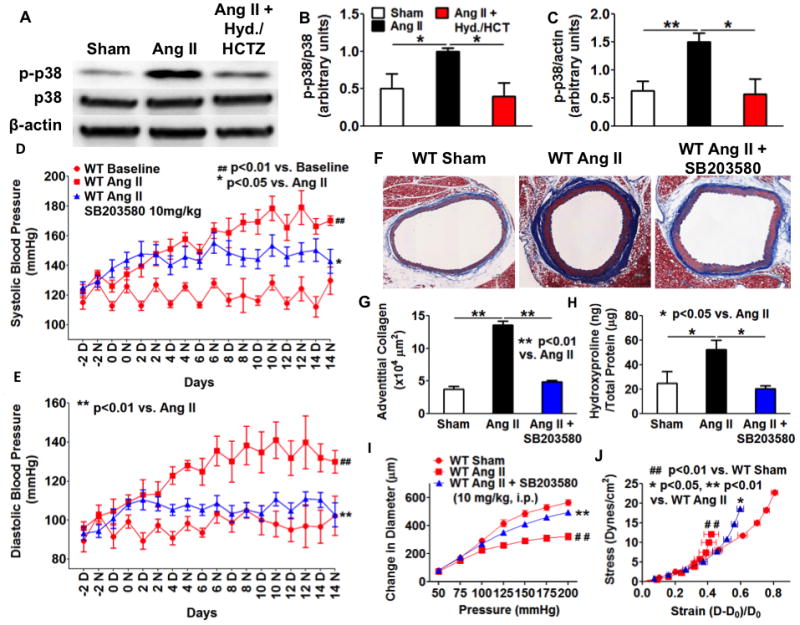
Chronic angiotensin II infusion caused marked aortic adventitial collagen deposition, as quantified by Masson trichrome blue staining and biochemically by hydroxyproline content, in wild-type but not in recombination activating gene-1-deficient mice. Aortic compliance, defined by ex vivo measurements of stress-strain curves, was reduced by chronic angiotensin II infusion in wild-type mice (P<0.01) but not in recombination activating gene-1-deficient mice (P<0.05). Adoptive transfer of T-cells to recombination activating gene-1-deficient mice restored aortic collagen deposition and stiffness to values observed in wild-type mice. Mice lacking the T-cell-derived cytokine interleukin 17a were also protected against aortic stiffening. In additional studies, we found that blood pressure normalization by treatment with hydralazine and hydrochlorothiazide prevented angiotensin II-induced vascular T-cell infiltration, aortic stiffening, and collagen deposition. Finally, we found that mechanical stretch induces the expression of collagen 1α1, 3α1, and 5a1 in cultured aortic fibroblasts in a p38 mitogen-activated protein kinase-dependent fashion, and that inhibition of p38 prevented angiotensin II-induced aortic stiffening in vivo. Interleukin 17a also induced collagen 3a1 expression via the activation of p38 mitogen-activated protein kinase.
Our data define a pathway in which inflammation and mechanical stretch lead to vascular inflammation that promotes collagen deposition. The resultant increase in aortic stiffness likely further worsens systolic hypertension and its attendant end-organ damage.
在高血压中,主动脉变硬通常会发生,并进一步使收缩压升高。高血压也与血管炎症和机械拉伸增加有关。在高血压中,炎症、机械拉伸和主动脉变硬之间的相互作用尚不清楚。
我们的目的是确定炎症和机械拉伸在主动脉变硬中的作用。
慢性血管紧张素 II 输注导致野生型小鼠主动脉外膜胶原沉积明显,通过 Masson 三色蓝染色和羟脯氨酸含量的生化方法进行定量;而重组激活基因-1 缺陷型小鼠则没有。在野生型小鼠中,慢性血管紧张素 II 输注导致主动脉顺应性(通过离体测量的应力-应变曲线定义)降低(P<0.01),但在重组激活基因-1 缺陷型小鼠中则没有(P<0.05)。将 T 细胞过继转移至重组激活基因-1 缺陷型小鼠,可使主动脉胶原沉积和硬度恢复至野生型小鼠观察到的水平。缺乏 T 细胞衍生细胞因子白细胞介素 17a 的小鼠也能防止主动脉变硬。在其他研究中,我们发现用肼屈嗪和氢氯噻嗪治疗可使血压正常化,从而防止血管紧张素 II 引起的血管 T 细胞浸润、主动脉变硬和胶原沉积。最后,我们发现机械拉伸以依赖 p38 丝裂原激活蛋白激酶的方式诱导培养的主动脉成纤维细胞中胶原 1α1、3α1 和 5a1 的表达,并且抑制 p38 可防止血管紧张素 II 诱导的体内主动脉变硬。白细胞介素 17a 也通过激活 p38 丝裂原激活蛋白激酶诱导胶原 3α1 的表达。
我们的数据定义了一条途径,其中炎症和机械拉伸导致血管炎症,从而促进胶原沉积。由此引起的主动脉硬度增加可能进一步使收缩压升高及其伴随的靶器官损伤恶化。