Kennedy Scott R, Schmitt Michael W, Fox Edward J, Kohrn Brendan F, Salk Jesse J, Ahn Eun Hyun, Prindle Marc J, Kuong Kawai J, Shen Jiang-Cheng, Risques Rosa-Ana, Loeb Lawrence A
Department of Pathology, University of Washington, Seattle, USA.
Department of Medicine, University of Washington, Seattle, USA.
Nat Protoc. 2014 Nov;9(11):2586-606. doi: 10.1038/nprot.2014.170. Epub 2014 Oct 9.
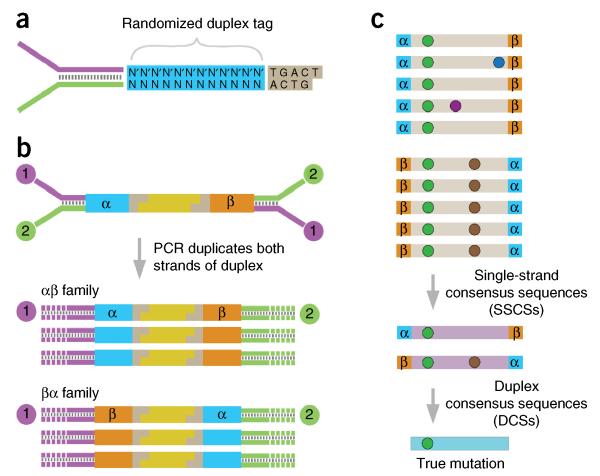
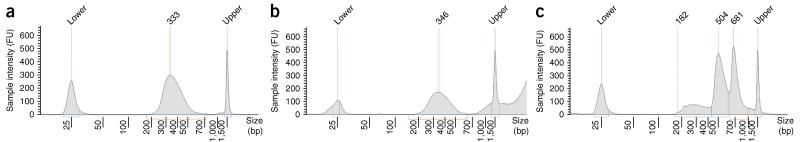
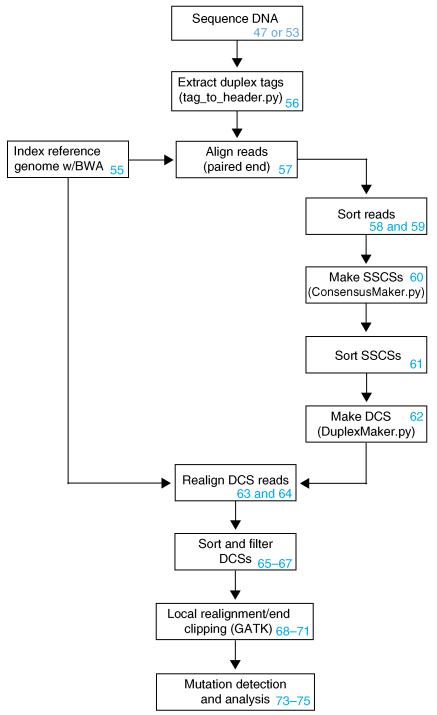
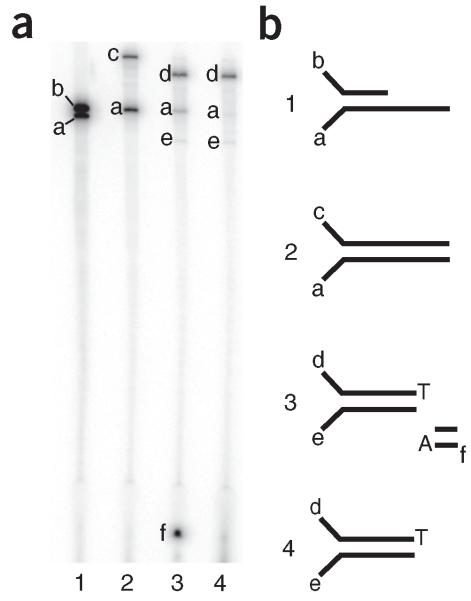
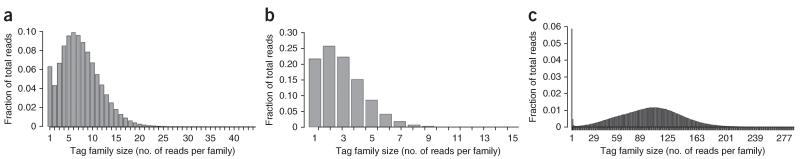
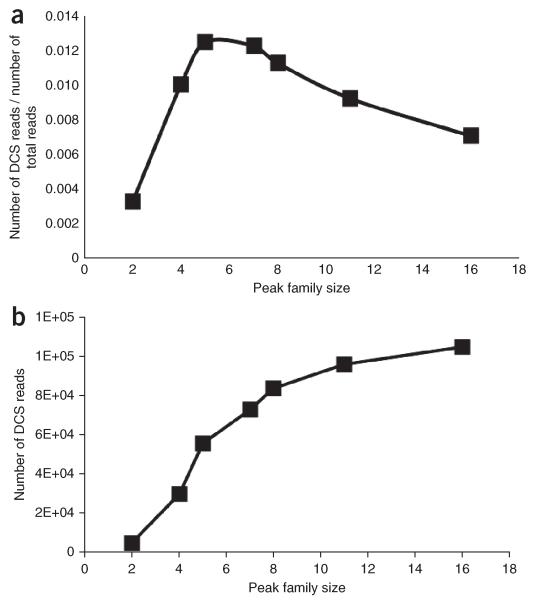
Duplex Sequencing (DS) is a next-generation sequencing methodology capable of detecting a single mutation among >1 × 10(7) wild-type nucleotides, thereby enabling the study of heterogeneous populations and very-low-frequency genetic alterations. DS can be applied to any double-stranded DNA sample, but it is ideal for small genomic regions of <1 Mb in size. The method relies on the ligation of sequencing adapters harboring random yet complementary double-stranded nucleotide sequences to the sample DNA of interest. Individually labeled strands are then PCR-amplified, creating sequence 'families' that share a common tag sequence derived from the two original complementary strands. Mutations are scored only if the variant is present in the PCR families arising from both of the two DNA strands. Here we provide a detailed protocol for efficient DS adapter synthesis, library preparation and target enrichment, as well as an overview of the data analysis workflow. The protocol typically takes 1-3 d.
双链测序(DS)是一种新一代测序方法,能够在超过1×10⁷个野生型核苷酸中检测到单个突变,从而能够研究异质群体和极低频率的基因改变。DS可应用于任何双链DNA样本,但它最适用于大小小于1 Mb的小基因组区域。该方法依赖于将携带随机但互补的双链核苷酸序列的测序接头连接到感兴趣的样本DNA上。然后对单独标记的链进行PCR扩增,创建共享源自两条原始互补链的共同标签序列的序列“家族”。只有当变体存在于由两条DNA链产生的PCR家族中时,才对突变进行评分。在这里,我们提供了一个详细的方案,用于高效的DS接头合成、文库制备和目标富集,以及数据分析工作流程的概述。该方案通常需要1 - 3天。