Touchon Marie, Cury Jean, Yoon Eun-Jeong, Krizova Lenka, Cerqueira Gustavo C, Murphy Cheryl, Feldgarden Michael, Wortman Jennifer, Clermont Dominique, Lambert Thierry, Grillot-Courvalin Catherine, Nemec Alexandr, Courvalin Patrice, Rocha Eduardo P C
Microbial Evolutionary Genomics, Institut Pasteur, Paris, France CNRS, UMR3525, Paris, France.
Unité des Agents Antibactériens, Institut Pasteur, Paris, France.
Genome Biol Evol. 2014 Oct 13;6(10):2866-82. doi: 10.1093/gbe/evu225.
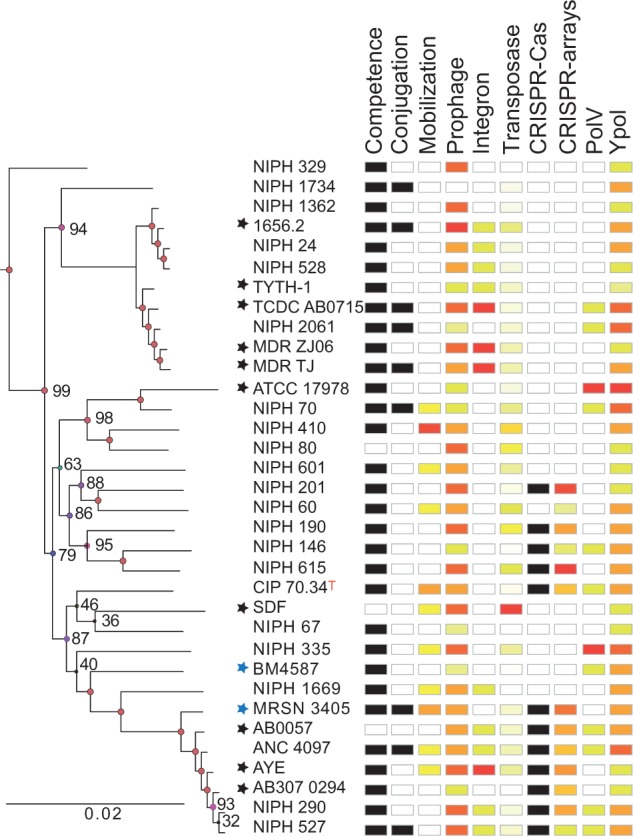
Bacterial genomics has greatly expanded our understanding of microdiversification patterns within a species, but analyses at higher taxonomical levels are necessary to understand and predict the independent rise of pathogens in a genus. We have sampled, sequenced, and assessed the diversity of genomes of validly named and tentative species of the Acinetobacter genus, a clade including major nosocomial pathogens and biotechnologically important species. We inferred a robust global phylogeny and delimited several new putative species. The genus is very ancient and extremely diverse: Genomes of highly divergent species share more orthologs than certain strains within a species. We systematically characterized elements and mechanisms driving genome diversification, such as conjugative elements, insertion sequences, and natural transformation. We found many error-prone polymerases that may play a role in resistance to toxins, antibiotics, and in the generation of genetic variation. Surprisingly, temperate phages, poorly studied in Acinetobacter, were found to account for a significant fraction of most genomes. Accordingly, many genomes encode clustered regularly interspaced short palindromic repeats (CRISPR)-Cas systems with some of the largest CRISPR-arrays found so far in bacteria. Integrons are strongly overrepresented in Acinetobacter baumannii, which correlates with its frequent resistance to antibiotics. Our data suggest that A. baumannii arose from an ancient population bottleneck followed by population expansion under strong purifying selection. The outstanding diversification of the species occurred largely by horizontal transfer, including some allelic recombination, at specific hotspots preferentially located close to the replication terminus. Our work sets a quantitative basis to understand the diversification of Acinetobacter into emerging resistant and versatile pathogens.
细菌基因组学极大地扩展了我们对物种内微观多样化模式的理解,但在更高分类水平上进行分析对于理解和预测一个属内病原体的独立出现是必要的。我们对不动杆菌属中有效命名和暂定物种的基因组进行了采样、测序和多样性评估,该进化枝包括主要的医院病原体和具有重要生物技术意义的物种。我们推断出一个可靠的全球系统发育树,并界定了几个新的假定物种。该属非常古老且极其多样:高度分化物种的基因组共享的直系同源基因比同一物种内的某些菌株更多。我们系统地描述了驱动基因组多样化的元件和机制,如接合元件、插入序列和自然转化。我们发现许多易出错的聚合酶可能在抗毒素、抗生素以及遗传变异产生中发挥作用。令人惊讶的是,在不动杆菌中研究较少的温和噬菌体在大多数基因组中占很大比例。因此,许多基因组编码成簇规律间隔短回文重复序列(CRISPR)-Cas系统,其中一些是迄今为止在细菌中发现的最大的CRISPR阵列。整合子在鲍曼不动杆菌中强烈富集,这与其频繁的抗生素耐药性相关。我们的数据表明,鲍曼不动杆菌起源于一个古老的种群瓶颈,随后在强烈的纯化选择下种群扩张。该物种的显著多样化主要通过水平转移发生,包括一些等位基因重组,发生在优先靠近复制终点的特定热点区域。我们的工作为理解不动杆菌向新兴耐药和多功能病原体的多样化提供了定量基础。