Department of Molecular Biology, Centro de Biología Molecular "Severo Ochoa," Consejo Superior de Investigaciones Científicas (CSIC), Universidad Autónoma de Madrid (UAM), Centro Investigación Biomédica en Red Enfermedades Neurodegenerativa (CIBERNED), Madrid, Spain.
Front Mol Neurosci. 2014 Sep 29;7:77. doi: 10.3389/fnmol.2014.00077. eCollection 2014.
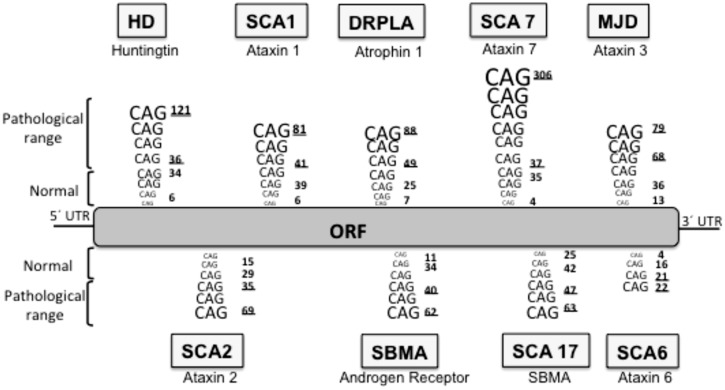
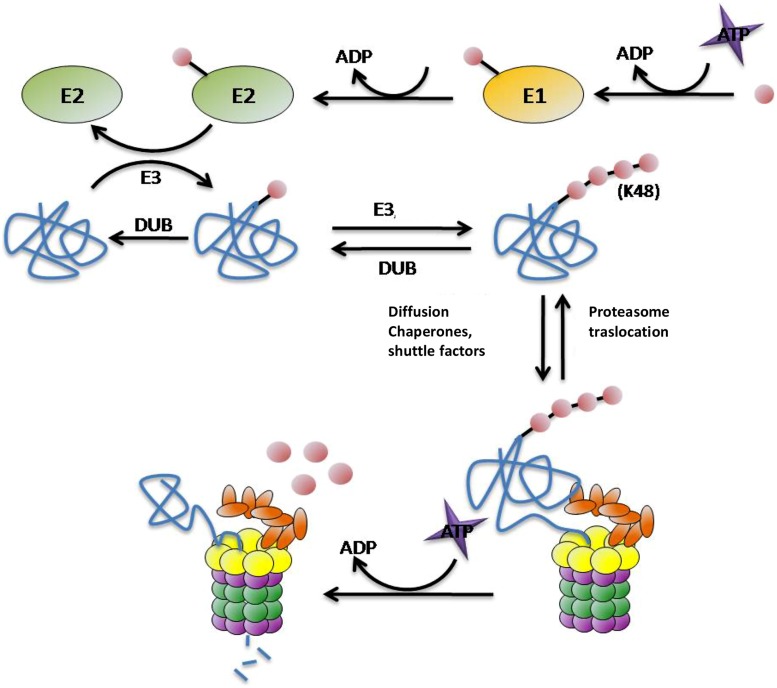
Huntington's disease (HD) is a genetic autosomal dominant neurodegenerative disease caused by the expansion of a CAG repeat in the huntingtin (htt) gene. This triplet expansion encodes a polyglutamine stretch (polyQ) in the N-terminus of the high molecular weight (348-kDa) and ubiquitously expressed protein htt. Normal individuals have between 6 and 35 CAG triplets, while expansions longer than 40 repeats lead to HD. The onset and severity of the disease depend on the length of the polyQ tract: the longer the polyglutamine stretch (polyQ) is, the earlier the disease begins and the more severe the symptoms are. One of the main histopathological hallmarks of HD is the presence of intraneuronal proteinaceous inclusion bodies, whose prominent and invariant feature is the presence of ubiquitin (Ub); therefore, they can be detected with anti-ubiquitin and anti-proteasome antibodies. This, together with the observation that mutations in components of the ubiquitin-proteasome system (UPS) give rise to some neurodegenerative diseases, suggests that UPS impairment may be causative of HD. Even though the link between disrupted Ub homeostasis and protein aggregation to HD is undisputed, the functional significance of these correlations and their mechanistic implications remains unresolved. Moreover, there is no consistent evidence documenting an accompanying decrease in levels of free Ub or disruption of Ub pool dynamics in neurodegenerative disease or models thus suggesting that the Ub-conjugate accumulation may be benign and just underlie lesion in 26S function. In this chapter we will elaborate on the different studies that have been performed using different experimental approaches, in order to shed light to this matter.
亨廷顿病(HD)是一种由亨廷顿(htt)基因中 CAG 重复扩展引起的常染色体显性遗传神经退行性疾病。这种三核苷酸扩展在高分子量(348kDa)和广泛表达的 htt 蛋白的 N 端编码一段聚谷氨酰胺延伸(polyQ)。正常个体有 6 到 35 个 CAG 三核苷酸,而超过 40 个重复的扩展会导致 HD。疾病的发病和严重程度取决于 polyQ 片段的长度:polyQ 延伸越长,疾病开始得越早,症状越严重。HD 的主要组织病理学特征之一是存在神经元内蛋白包涵体,其突出和不变的特征是存在泛素(Ub);因此,可以用抗泛素和抗蛋白酶体抗体检测到它们。这一点,再加上观察到泛素-蛋白酶体系统(UPS)成分的突变会导致一些神经退行性疾病,表明 UPS 损伤可能是 HD 的原因。尽管 Ub 动态平衡破坏和蛋白质聚集与 HD 之间的联系是无可争议的,但这些相关性的功能意义及其机制意义仍未解决。此外,没有一致的证据证明在神经退行性疾病或模型中存在游离 Ub 水平降低或 Ub 池动力学紊乱,这表明 Ub 缀合物的积累可能是良性的,只是 26S 功能的病变基础。在本章中,我们将详细阐述使用不同实验方法进行的不同研究,以阐明这一问题。