Tian Lijun, Cao Junhua, Deng Xingqiang, Zhang Chuanling, Qian Tong, Song Xianxiang, Huang Baoshan
Clinical laboratory, Xuzhou Children's Hospital, No. 18 Sudibei Road, Xuzhou, Jiangsu Province, China.
Diagn Pathol. 2014 Oct 23;9:210. doi: 10.1186/s13000-014-0210-z.
Gene expression analysis is powerful for investigating the underlying mechanisms of Duchenne muscular dystrophy (DMD). Previous studies mainly neglected co-expression or transcription factor (TF) information. Here we integrated TF information into differential co-expression analysis (DCEA) to explore new understandings of DMD pathogenesis.
Using two microarray datasets from Gene Expression Omnibus (GEO) database, we firstly detected differentially expressed genes (DEGs) and pathways enriched with DEGs. Secondly, we constructed differentially regulated networks to integrate the TF-to-target information and the differential co-expression genes.
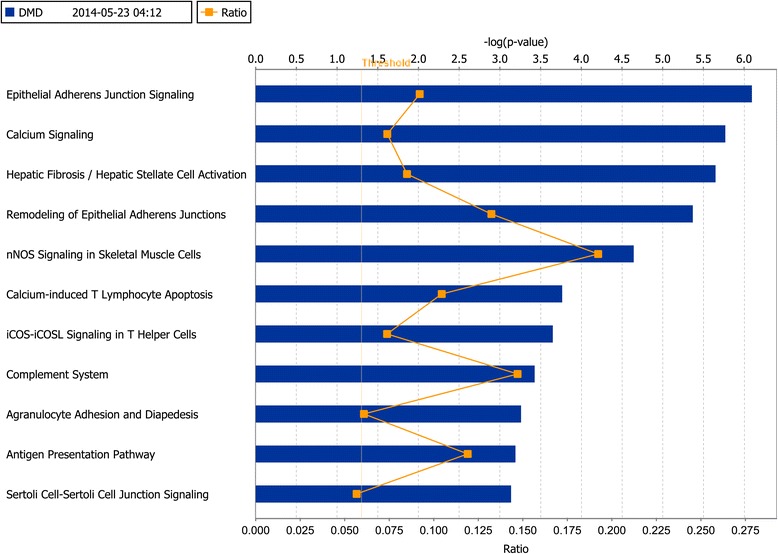
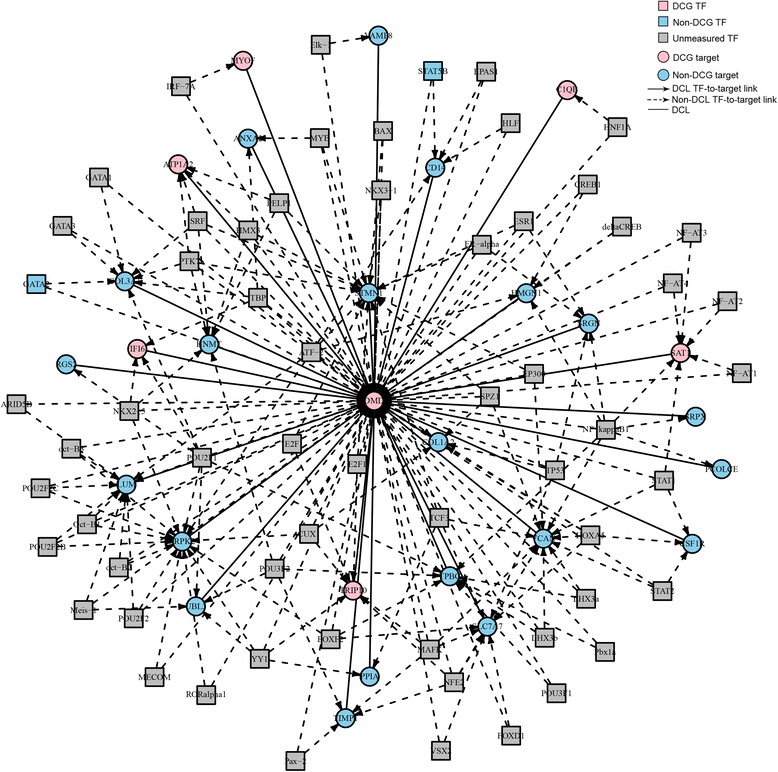
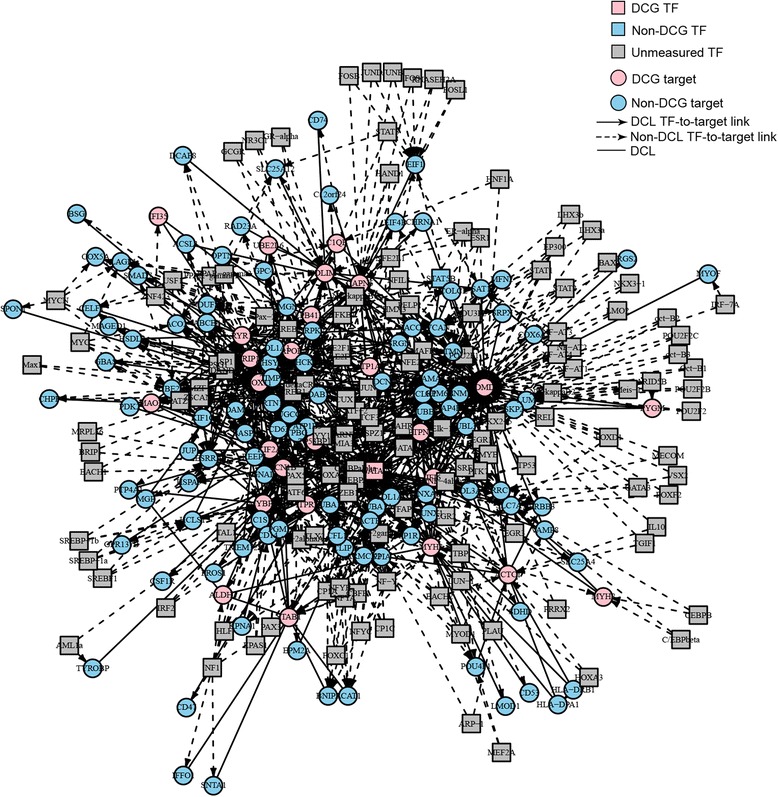
A total of 454 DEGs were detected and both KEGG pathway and ingenuity pathway analysis revealed that pathways enriched with aberrantly regulated genes are mostly involved in the immune response processes. DCEA results generated 610 pairs of DEGs regulated by at least one common TF, including 78 pairs of co-expressed DEGs. A network was constructed to illustrate their relationships and a subnetwork for DMD related molecules was constructed to show genes and TFs that may play important roles in the secondary changes of DMD. Among the DEGs which shared TFs with DMD, six genes were co-expressed with DMD, including ATP1A2, C1QB, MYOF, SAT1, TRIP10, and IFI6.
Our results may provide a new understanding of DMD and contribute potential targets for future therapeutic tests.
The virtual slide(s) for this article can be found here: http://www.diagnosticpathology.diagnomx.eu/vs/13000_2014_210.
基因表达分析对于研究杜氏肌营养不良症(DMD)的潜在机制具有重要作用。以往的研究主要忽略了共表达或转录因子(TF)信息。在此,我们将TF信息整合到差异共表达分析(DCEA)中,以探索对DMD发病机制的新认识。
利用来自基因表达综合数据库(GEO)的两个微阵列数据集,我们首先检测差异表达基因(DEG)以及富含DEG的通路。其次,我们构建差异调控网络,以整合TF与靶标的信息以及差异共表达基因。
共检测到454个DEG,KEGG通路分析和 Ingenuity 通路分析均显示,富含异常调控基因的通路大多参与免疫应答过程。DCEA结果产生了610对受至少一个共同TF调控的DEG,其中包括78对共表达的DEG。构建了一个网络来说明它们之间的关系,并构建了一个与DMD相关分子的子网,以展示可能在DMD继发性变化中起重要作用的基因和TF。在与DMD共享TF的DEG中,有六个基因与DMD共表达,包括ATP1A2、C1QB、MYOF、SAT1、TRIP10和IFI6。
我们的结果可能为DMD提供新的认识,并为未来的治疗试验贡献潜在靶点。
本文的虚拟切片可在此处找到:http://www.diagnosticpathology.diagnomx.eu/vs/13000_2014_210。