Mukund Kavitha, Subramaniam Shankar
Bioinformatics and System Biology Graduate Program, University of California San Diego, 9500 Gilman Drive, MC0412, La Jolla, CA, 92093, USA.
Departments of Bioengineering, Computer Science & Engineering, Cellular & Molecular Medicine and Chemistry & Biochemistry University of California, San Diego, 9500 Gilman Drive, MC0412, La Jolla, CA, 92093, USA.
BMC Res Notes. 2015 May 3;8:182. doi: 10.1186/s13104-015-1141-9.
Duchenne Muscular Dystrophy (DMD) is an X-linked recessive disorder with its primary insult on the skeletal muscle. Severe muscle wasting, chronic inflammation and fibrosis characterize dystrophic muscle. Here we identify dysregulated pathways in DMD utilizing a co-expression network approach as described in Weighted Gene Co-expression Network Analysis (WGCNA). Specifically, we utilize WGCNA's "preservation" statistics to identify gene modules that exhibit a weak conservation of network topology within healthy and dystrophic networks. Preservation statistics rank modules based on their topological metrics such as node density, connectivity and separability between networks.
Raw data for DMD was downloaded from Gene Expression Omnibus (GSE6011) and suitably preprocessed. Co-expression networks for each condition (healthy and dystrophic) were generated using the WGCNA library in R. Preservation of healthy network edges was evaluated with respect to dystrophic muscle and vice versa using WGCNA. Highly exclusive gene pairs for each of the low preserved modules within both networks were also determined using a specificity measure.
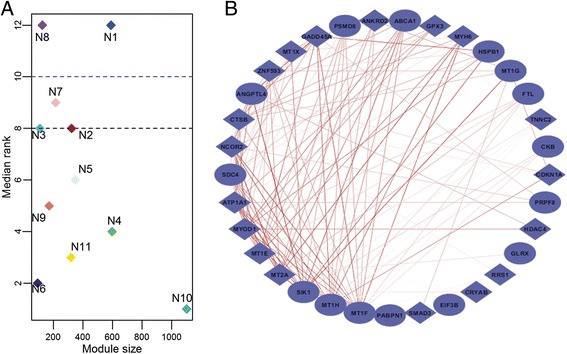
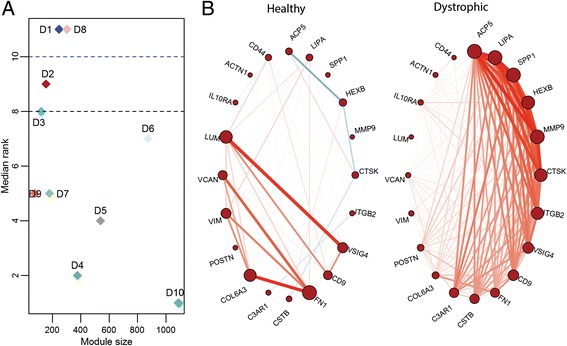
A total of 11 and 10 co-expressed modules were identified in the networks generated from 13 healthy and 23 dystrophic samples respectively. 5 out of the 11, and 4 out of the 10 modules were identified as exhibiting none-to-weak preservation. Functional enrichment analysis identified that these weakly preserved modules were highly relevant to the condition under study. For instance, weakly preserved dystrophic module D2 exhibited the highest fraction of genes exclusive to DMD. The highly specific gene pairs identified within these modules were enriched for genes activated in response to wounding and affect the extracellular matrix including several markers such as SPP1, MMP9 and ITGB2.
The proposed approach allowed us to identify clusters of genes that are non-randomly associated with the disease. Furthermore, highly specific gene pairs pointed to interactions between known markers of disease and identification of putative markers likely associated with disease. The analysis also helped identify putative novel interactions associated with the progression of DMD.
杜氏肌营养不良症(DMD)是一种X连锁隐性疾病,主要损害骨骼肌。严重的肌肉萎缩、慢性炎症和纤维化是营养不良性肌肉的特征。在这里,我们利用加权基因共表达网络分析(WGCNA)中描述的共表达网络方法,识别DMD中失调的通路。具体来说,我们利用WGCNA的“保守性”统计来识别在健康和营养不良网络中表现出网络拓扑结构弱保守性的基因模块。保守性统计根据模块的拓扑指标(如节点密度、连通性和网络之间的可分离性)对模块进行排名。
从基因表达综合数据库(GSE6011)下载DMD的原始数据并进行适当预处理。使用R中的WGCNA库生成每种情况(健康和营养不良)的共表达网络。使用WGCNA评估营养不良性肌肉相对于健康网络边的保守性,反之亦然。还使用特异性度量确定两个网络中每个低保守模块的高度排他性基因对。
分别从13个健康样本和23个营养不良样本生成的网络中,共识别出11个和10个共表达模块。11个模块中的5个和10个模块中的4个被确定为表现出无到弱的保守性。功能富集分析表明,这些弱保守模块与所研究的疾病高度相关。例如,弱保守的营养不良模块D2显示出DMD特有的基因比例最高。在这些模块中鉴定出的高度特异性基因对富含因受伤而激活并影响细胞外基质的基因,包括几种标志物,如SPP1、MMP9和ITGB2。
所提出的方法使我们能够识别与疾病非随机相关的基因簇。此外,高度特异性的基因对指向疾病已知标志物之间的相互作用,并识别可能与疾病相关的假定标志物。该分析还有助于识别与DMD进展相关的假定新相互作用。