Department of Medicine and the Moores UCSD Cancer Center, University of California, 3855 Health Sciences Drive, La Jolla, San Diego, CA, 92093-0819, USA.
Cell Commun Signal. 2014 Oct 25;12:59. doi: 10.1186/s12964-014-0059-5.
The EphA2 receptor, which is expressed in many types of cancer, is activated by two different mechanisms. Activation by engagement with one of its ephrin ligands is anti-oncogenic whereas phosphorylation of S897 by AKT increases migration, invasion and metastasis. Down-regulation of claudin-4 (CLDN4) produces a loss of E-cadherin and increased β-catenin signaling and a phenotype similar to that produced by oncogenic activation of EphA2, suggesting that CLDN4 may serve to restrain the pro-oncogenic signaling of EphA2.
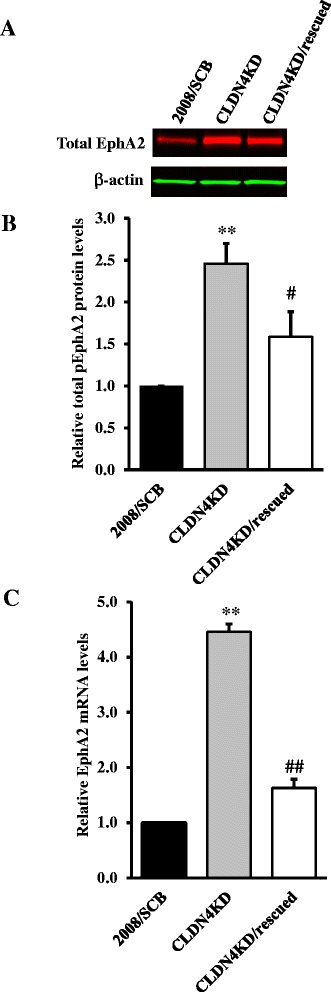
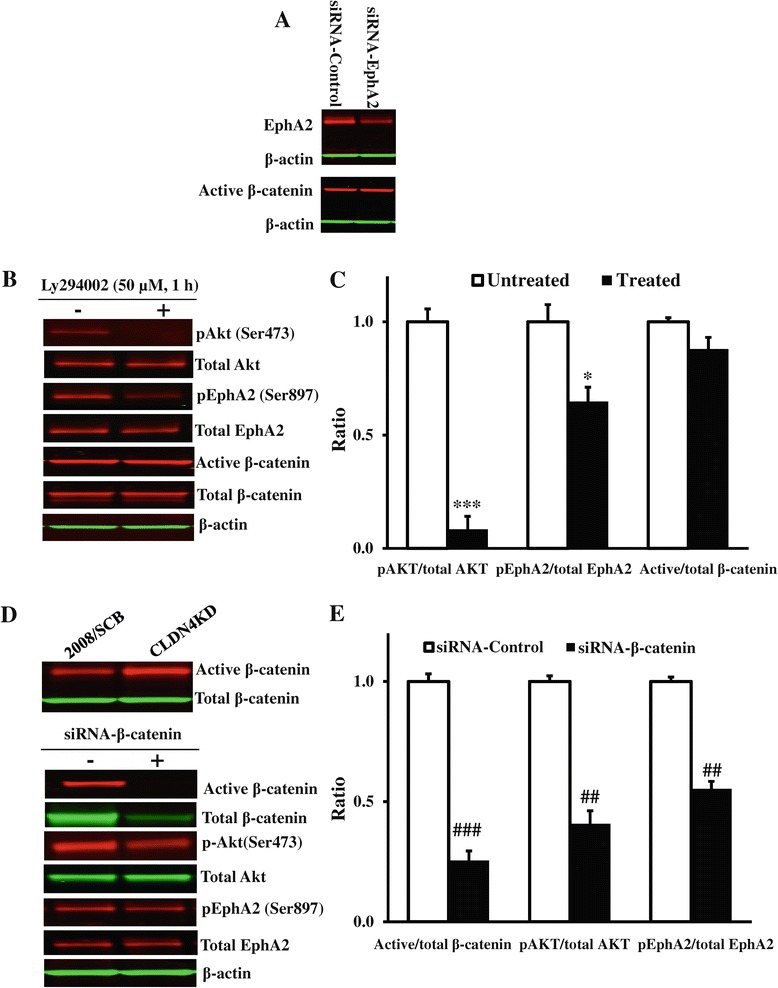
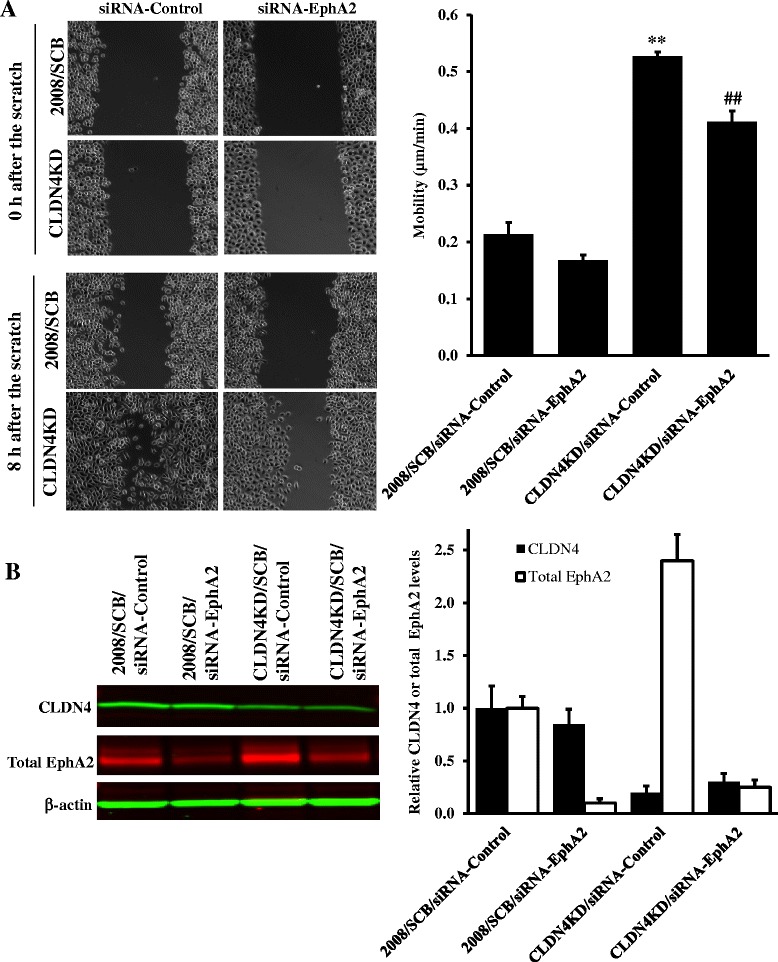
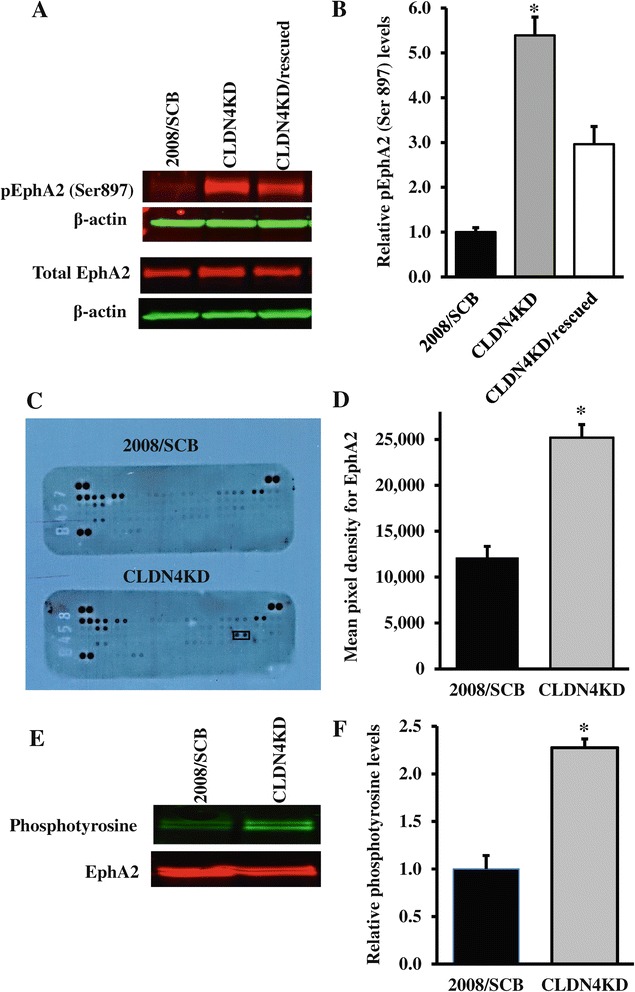
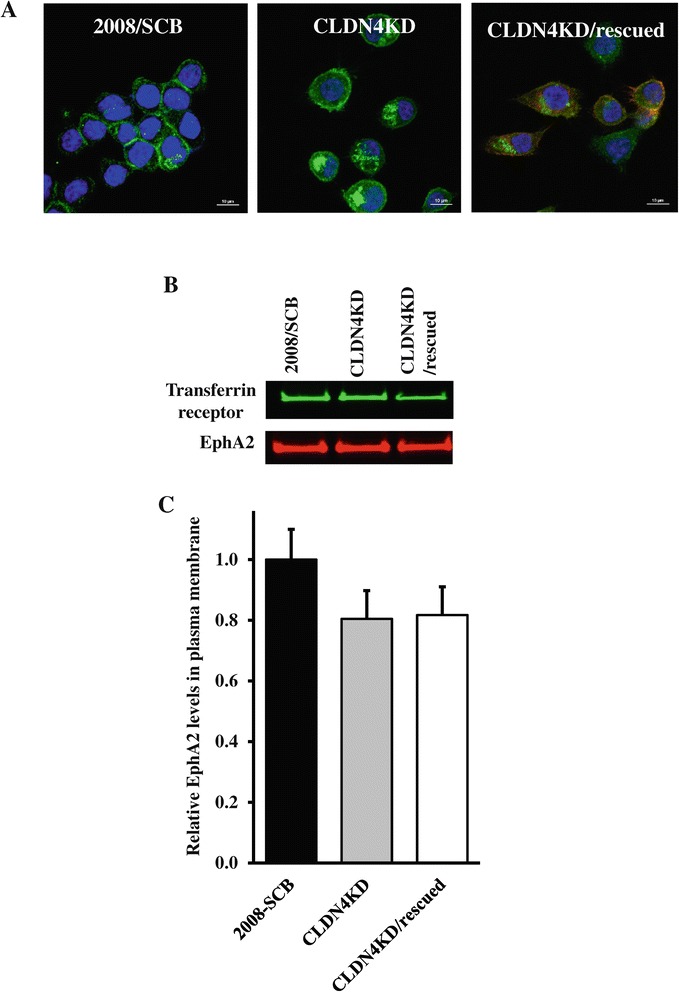
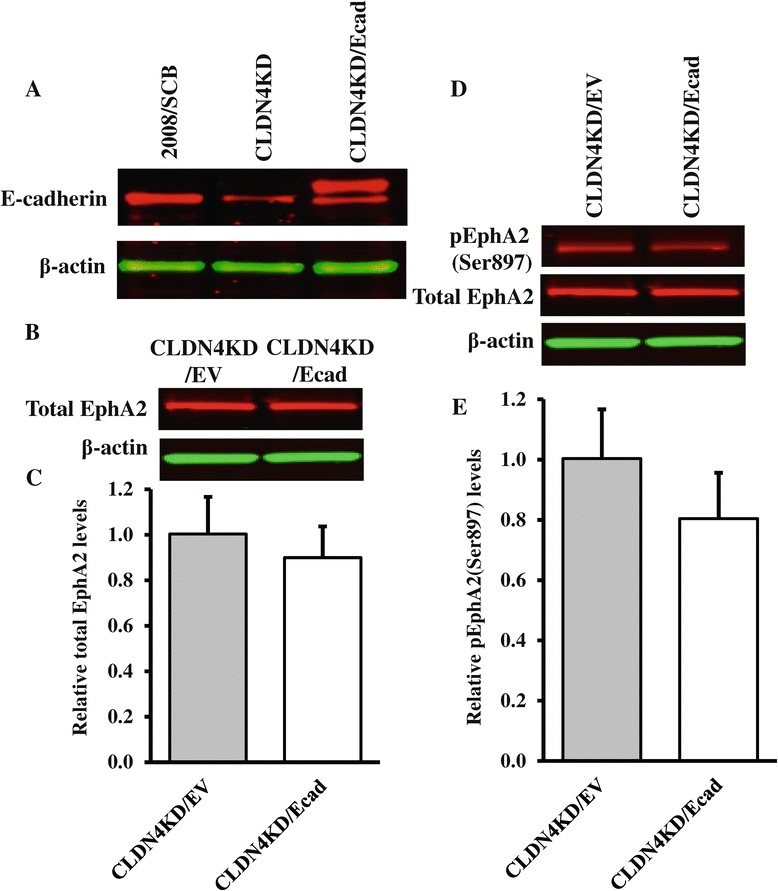
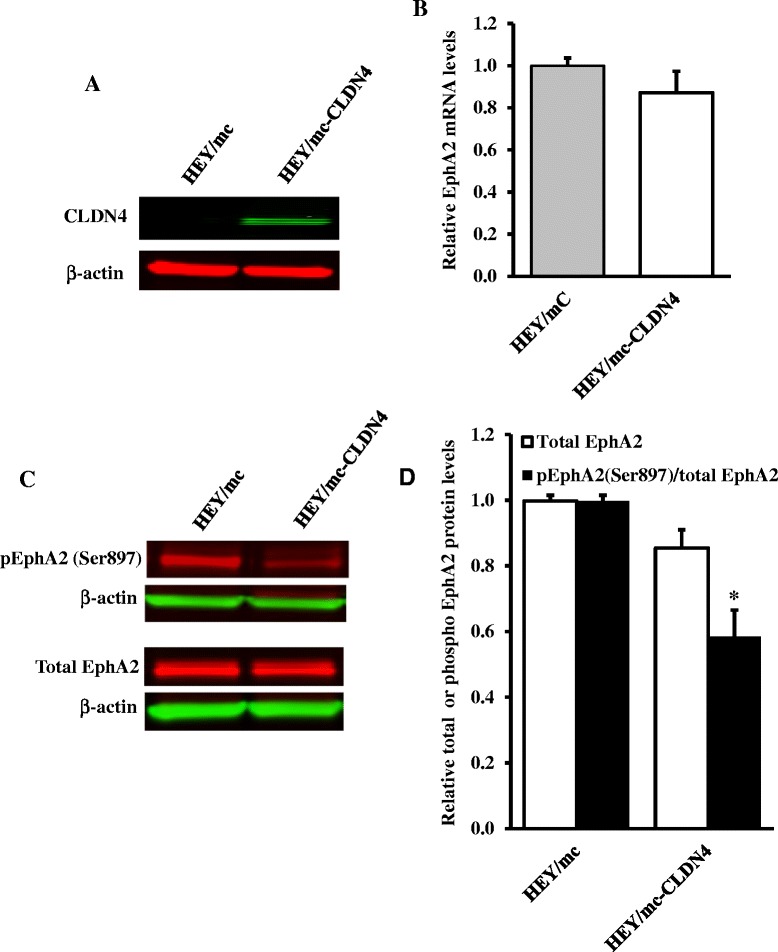
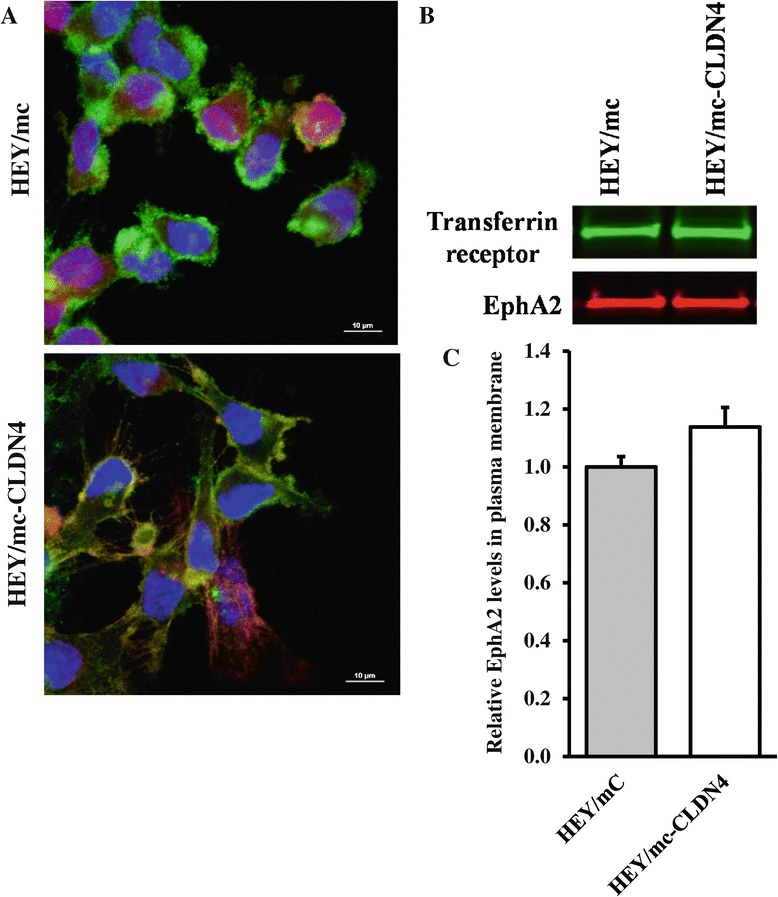
We found that constitutive knockdown of CLDN4 was associated with a 4.5-fold increase in EphA2 mRNA and a 2.5-fold increase in EphA2 protein which was reversible by re-expression of CLDN4. Knockdown of EphA2 blocked the migratory phenotype induced by loss of CLDN4. Knockdown of CLDN4 resulted in a 5.8-fold increase in pEphA(S897), the oncogenic form of the receptor, as well as partial mislocalization of the excess EphA2 to the interior of the cell. Forced expression of E-cadherin did not reduce total EphA2 or pEphA(S897) whereas re-expression of CLDN4 restored localization and reduced EphA2 and pEphA(S897) even in cells not expressing E-cadherin. Transient siRNA-mediated knockdown of EphA2 and β-catenin, and inhibition of PI3K by LY294002, demonstrated that increased pEphA(S897) in the CLDN4 knockdown cells was attributable to an increase in the level of active dephospho-β-catenin upstream of PI3K and AKT.
We conclude that CLDN4 serves to restrain pro-oncogenic signaling from EphA2 by limiting the activity of β-catenin and PI3K and preventing phosphorylation of EphA2 on S897 by AKT. This suggests that interventions directed at enhancing the level or functional activity of CLDN4 may be of therapeutic interest.
EphA2 受体在多种类型的癌症中表达,它有两种不同的激活机制。与其中一种 Ephrin 配体结合的激活是抗肿瘤的,而 AKT 磷酸化 S897 则会增加迁移、侵袭和转移。 Claudin-4 (CLDN4) 的下调会导致 E-钙粘蛋白的丢失和 β-连环蛋白信号的增加,以及类似于 EphA2 致癌激活所产生的表型,这表明 CLDN4 可能有助于抑制 EphA2 的促癌信号。
我们发现,CLDN4 的组成性敲低与 EphA2 mRNA 增加 4.5 倍和 EphA2 蛋白增加 2.5 倍有关,而 CLDN4 的重新表达可使其逆转。 EphA2 的敲低阻断了 CLDN4 缺失诱导的迁移表型。CLDN4 的敲低导致 pEphA(S897)(受体的致癌形式)增加 5.8 倍,以及过量 EphA2 的部分错误定位到细胞内部。强制表达 E-钙粘蛋白不会减少总 EphA2 或 pEphA(S897),而 CLDN4 的重新表达即使在不表达 E-钙粘蛋白的细胞中也能恢复定位并减少 EphA2 和 pEphA(S897)。 EphA2 和 β-连环蛋白的瞬时 siRNA 介导的敲低,以及 PI3K 的 LY294002 抑制,表明 CLDN4 敲低细胞中 pEphA(S897)的增加归因于 PI3K 和 AKT 上游活性去磷酸化 β-连环蛋白水平的增加。
我们得出结论,CLDN4 通过限制 β-连环蛋白和 PI3K 的活性以及防止 AKT 对 EphA2 的 S897 磷酸化,从而限制 EphA2 的促癌信号。这表明,针对增强 CLDN4 的水平或功能活性的干预可能具有治疗意义。