Martelotto Luciano G, Ng Charlotte Ky, De Filippo Maria R, Zhang Yan, Piscuoglio Salvatore, Lim Raymond S, Shen Ronglai, Norton Larry, Reis-Filho Jorge S, Weigelt Britta
Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA.
Genome Biol. 2014 Oct 28;15(10):484. doi: 10.1186/s13059-014-0484-1.
Massively parallel sequencing studies have led to the identification of a large number of mutations present in a minority of cancers of a given site. Hence, methods to identify the likely pathogenic mutations that are worth exploring experimentally and clinically are required. We sought to compare the performance of 15 mutation effect prediction algorithms and their agreement. As a hypothesis-generating aim, we sought to define whether combinations of prediction algorithms would improve the functional effect predictions of specific mutations.
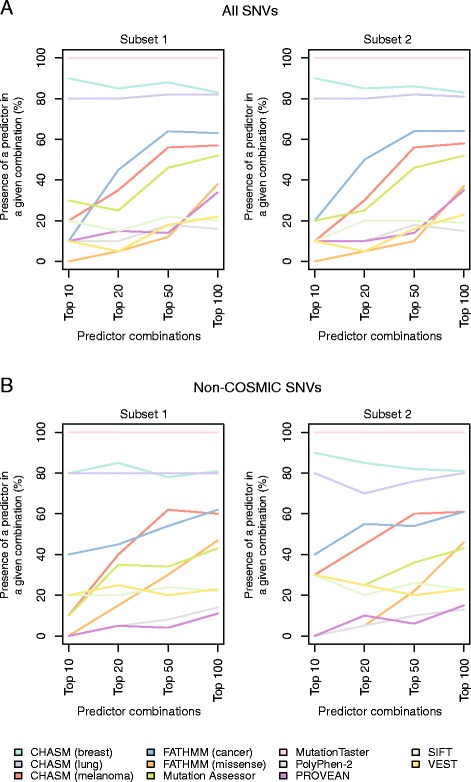
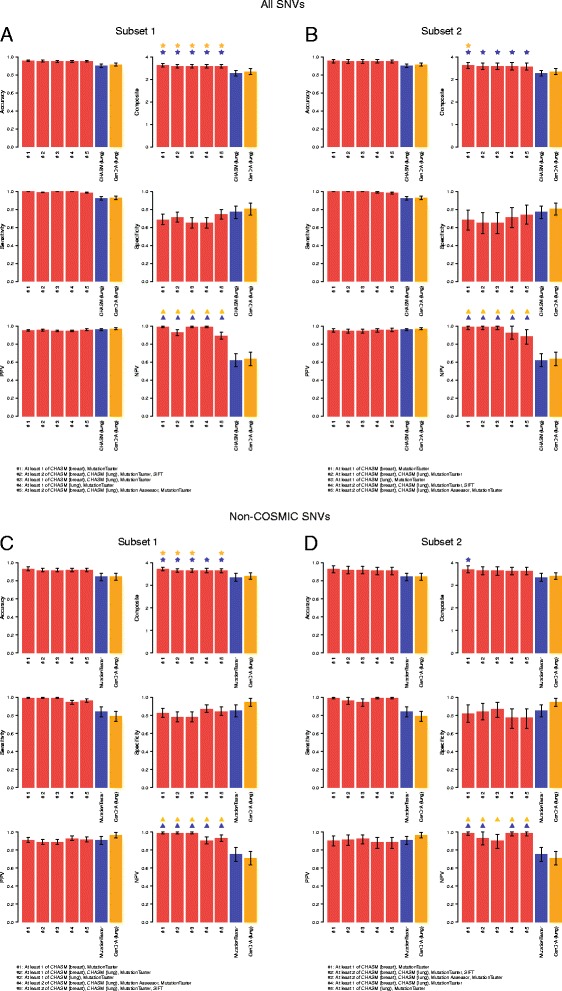
Literature and database mining of single nucleotide variants (SNVs) affecting 15 cancer genes was performed to identify mutations supported by functional evidence or hereditary disease association to be classified either as non-neutral (n = 849) or neutral (n = 140) with respect to their impact on protein function. These SNVs were employed to test the performance of 15 mutation effect prediction algorithms. The accuracy of the prediction algorithms varies considerably. Although all algorithms perform consistently well in terms of positive predictive value, their negative predictive value varies substantially. Cancer-specific mutation effect predictors display no-to-almost perfect agreement in their predictions of these SNVs, whereas the non-cancer-specific predictors showed no-to-moderate agreement. Combinations of predictors modestly improve accuracy and significantly improve negative predictive values.
The information provided by mutation effect predictors is not equivalent. No algorithm is able to predict sufficiently accurately SNVs that should be taken forward for experimental or clinical testing. Combining algorithms aggregates orthogonal information and may result in improvements in the negative predictive value of mutation effect predictions.
大规模平行测序研究已导致在特定部位的少数癌症中鉴定出大量突变。因此,需要有方法来鉴定那些值得在实验和临床中探索的可能致病突变。我们试图比较15种突变效应预测算法的性能及其一致性。作为一个产生假设的目标,我们试图确定预测算法的组合是否会改善特定突变的功能效应预测。
对影响15个癌症基因的单核苷酸变异(SNV)进行文献和数据库挖掘,以鉴定得到功能证据或遗传性疾病关联支持的突变,根据其对蛋白质功能的影响将其分类为非中性(n = 849)或中性(n = 140)。这些SNV被用于测试15种突变效应预测算法的性能。预测算法的准确性差异很大。尽管所有算法在阳性预测值方面表现一致良好,但其阴性预测值差异很大。癌症特异性突变效应预测器在对这些SNV的预测中显示出从无到几乎完美的一致性,而非癌症特异性预测器则显示出从无到中等的一致性。预测器的组合适度提高了准确性,并显著提高了阴性预测值。
突变效应预测器提供的信息并不等同。没有一种算法能够足够准确地预测哪些SNV应该进行实验或临床测试。组合算法汇总了正交信息,并可能导致突变效应预测的阴性预测值得到改善。