Department of Internal Medicine, Fundación de Investigación Biomédica, Hospital Universitario de La Princesa, Madrid, Spain.
Clin Epidemiol. 2014 Oct 29;6:369-77. doi: 10.2147/CLEP.S39981. eCollection 2014.
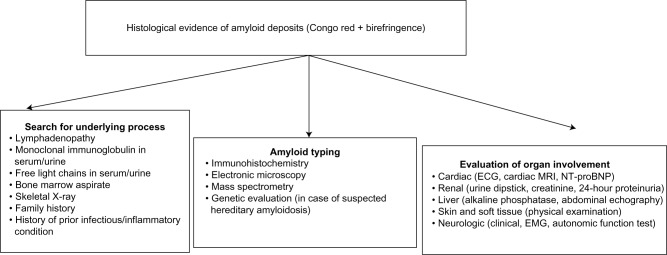
The term "amyloidosis" encompasses the heterogeneous group of diseases caused by the extracellular deposition of autologous fibrillar proteins. The global incidence of amyloidosis is estimated at five to nine cases per million patient-years. While amyloid light-chain (AL) amyloidosis is more frequent in developed countries, amyloid A (AA) amyloidosis is more common in some European regions and in developing countries. The spectrum of AA amyloidosis has changed in recent decades owing to: an increase in the median age at diagnosis; a percent increase in the frequency of primary AL amyloidosis with respect to the AA type; and a substantial change in the epidemiology of the underlying diseases. Diagnosis of amyloidosis is based on clinical organ involvement and histological evidence of amyloid deposits. Among the many tinctorial characteristics of amyloid deposits, avidity for Congo red and metachromatic birefringence under unidirectional polarized light remain the gold standard. Once the initial diagnosis has been made, the amyloid subtype must be identified and systemic organ involvement evaluated. In this sense, the (123)I-labeled serum amyloid P component scintigraphy is a safe and noninvasive technique that has revolutionized the diagnosis and monitoring of treatment in systemic amyloidosis. It can successfully identify anatomical patterns of amyloid deposition throughout the body and enables not only an initial estimation of prognosis, but also the monitoring of the course of the disease and the response to treatment. Given the etiologic diversity of AA amyloidosis, common therapeutic strategies are scarce. All treatment options should be based upon a greater control of the underlying disease, adequate organ support, and treatment of symptoms. Nevertheless, novel therapeutic strategies targeting the formation of amyloid fibrils and amyloid deposition may generate new expectations for patients with AA amyloidosis.
“淀粉样变性”一词涵盖了由自体纤维状蛋白细胞外沉积引起的异质性疾病群体。淀粉样变性的全球发病率估计为每百万患者年五至九例。虽然轻链淀粉样变性(AL)在发达国家更为常见,但淀粉样 A(AA)淀粉样变性在一些欧洲地区和发展中国家更为常见。由于以下原因,AA 淀粉样变性的谱在最近几十年发生了变化:诊断时的中位年龄增加;与 AA 型相比,原发性 AL 淀粉样变性的频率增加;以及潜在疾病的流行病学发生了实质性变化。淀粉样变性的诊断基于临床器官受累和组织学证据的淀粉样沉积物。在淀粉样沉积物的许多染色特征中,刚果红的亲和力和单方向偏振光下的变色双折射仍然是金标准。一旦做出初步诊断,就必须确定淀粉样变亚型并评估系统性器官受累。在这种意义上,(123)I 标记血清淀粉样蛋白 P 成分闪烁扫描是一种安全且非侵入性的技术,它彻底改变了系统性淀粉样变性的诊断和治疗监测。它可以成功识别全身淀粉样沉积的解剖模式,不仅可以初步估计预后,还可以监测疾病的过程和对治疗的反应。鉴于 AA 淀粉样变性的病因多样性,常见的治疗策略很少。所有治疗选择都应基于更好地控制基础疾病、充分的器官支持和症状治疗。然而,针对淀粉样纤维形成和淀粉样沉积的新型治疗策略可能为 AA 淀粉样变性患者带来新的希望。