Halachev Mihail R, Chan Jacqueline Z-M, Constantinidou Chrystala I, Cumley Nicola, Bradley Craig, Smith-Banks Matthew, Oppenheim Beryl, Pallen Mark J
Institute of Microbiology and Infection, University of Birmingham, Birmingham, B15 2TT UK.
Division of Microbiology and Infection, Warwick Medical School, University of Warwick, Warwick, CV4 7AL UK.
Genome Med. 2014 Nov 20;6(11):70. doi: 10.1186/s13073-014-0070-x. eCollection 2014.
Multidrug-resistant Acinetobacter baumannii commonly causes hospital outbreaks. However, within an outbreak, it can be difficult to identify the routes of cross-infection rapidly and accurately enough to inform infection control. Here, we describe a protracted hospital outbreak of multidrug-resistant A. baumannii, in which whole-genome sequencing (WGS) was used to obtain a high-resolution view of the relationships between isolates.
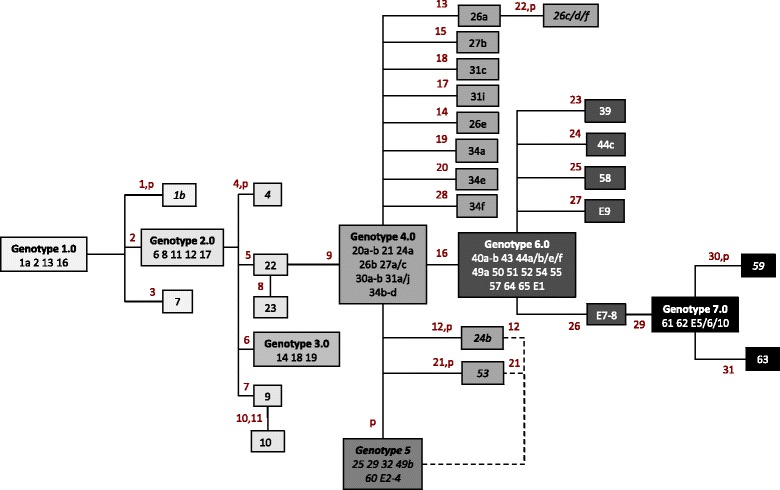
To delineate and investigate the outbreak, we attempted to genome-sequence 114 isolates that had been assigned to the A. baumannii complex by the Vitek2 system and obtained informative draft genome sequences from 102 of them. Genomes were mapped against an outbreak reference sequence to identify single nucleotide variants (SNVs).
We found that the pulsotype 27 outbreak strain was distinct from all other genome-sequenced strains. Seventy-four isolates from 49 patients could be assigned to the pulsotype 27 outbreak on the basis of genomic similarity, while WGS allowed 18 isolates to be ruled out of the outbreak. Among the pulsotype 27 outbreak isolates, we identified 31 SNVs and seven major genotypic clusters. In two patients, we documented within-host diversity, including mixtures of unrelated strains and within-strain clouds of SNV diversity. By combining WGS and epidemiological data, we reconstructed potential transmission events that linked all but 10 of the patients and confirmed links between clinical and environmental isolates. Identification of a contaminated bed and a burns theatre as sources of transmission led to enhanced environmental decontamination procedures.
WGS is now poised to make an impact on hospital infection prevention and control, delivering cost-effective identification of routes of infection within a clinically relevant timeframe and allowing infection control teams to track, and even prevent, the spread of drug-resistant hospital pathogens.
多重耐药鲍曼不动杆菌常引发医院感染暴发。然而,在一次暴发中,要快速、准确地识别交叉感染途径以指导感染控制可能具有挑战性。在此,我们描述了一次长期的多重耐药鲍曼不动杆菌医院感染暴发,其中使用全基因组测序(WGS)来深入了解分离株之间的关系。
为了描绘和调查此次暴发,我们尝试对114株经Vitek2系统鉴定为鲍曼不动杆菌复合体的分离株进行基因组测序,并从其中102株获得了信息丰富的基因组草图序列。将基因组与暴发参考序列进行比对以识别单核苷酸变异(SNV)。
我们发现脉冲型27暴发菌株与所有其他经基因组测序的菌株不同。基于基因组相似性,49名患者的74株分离株可归为脉冲型27暴发,而WGS排除了18株分离株与此次暴发的关联。在脉冲型27暴发分离株中,我们鉴定出31个SNV和7个主要基因型簇。在两名患者中,我们记录了宿主内的多样性,包括不相关菌株的混合以及SNV多样性的菌株内云状分布。通过结合WGS和流行病学数据,我们重建了除10名患者外所有患者之间的潜在传播事件,并证实了临床分离株与环境分离株之间的联系。识别出一张受污染的病床和一个烧伤病房为传播源,从而加强了环境去污程序。
WGS目前有望对医院感染预防和控制产生影响,在临床相关时间范围内以具有成本效益的方式识别感染途径,并使感染控制团队能够追踪甚至预防耐药医院病原体的传播。