Jian Xueqiu, Boerwinkle Eric, Liu Xiaoming
Nucleic Acids Res. 2014 Dec 16;42(22):13534-44. doi: 10.1093/nar/gku1206.
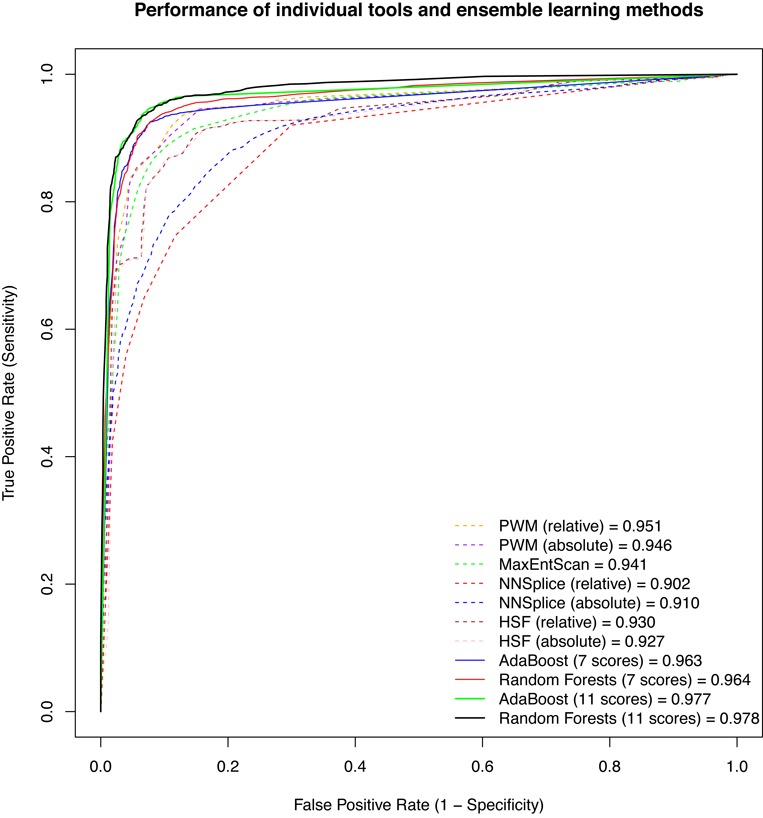
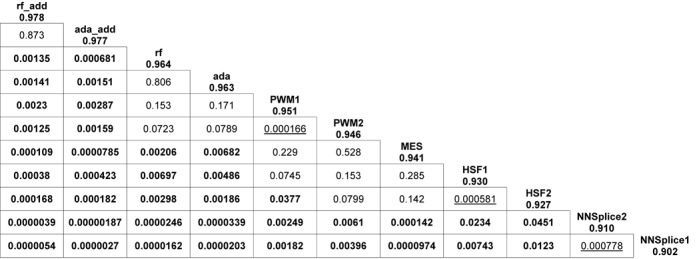
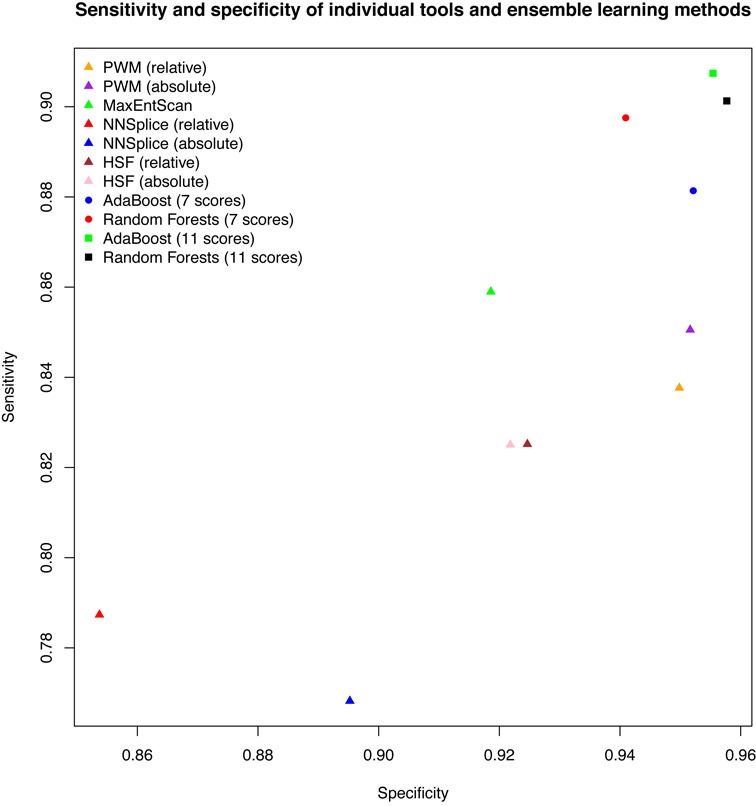
In silico tools have been developed to predict variants that may have an impact on pre-mRNA splicing. The major limitation of the application of these tools to basic research and clinical practice is the difficulty in interpreting the output. Most tools only predict potential splice sites given a DNA sequence without measuring splicing signal changes caused by a variant. Another limitation is the lack of large-scale evaluation studies of these tools. We compared eight in silico tools on 2959 single nucleotide variants within splicing consensus regions (scSNVs) using receiver operating characteristic analysis. The Position Weight Matrix model and MaxEntScan outperformed other methods. Two ensemble learning methods, adaptive boosting and random forests, were used to construct models that take advantage of individual methods. Both models further improved prediction, with outputs of directly interpretable prediction scores. We applied our ensemble scores to scSNVs from the Catalogue of Somatic Mutations in Cancer database. Analysis showed that predicted splice-altering scSNVs are enriched in recurrent scSNVs and known cancer genes. We pre-computed our ensemble scores for all potential scSNVs across the human genome, providing a whole genome level resource for identifying splice-altering scSNVs discovered from large-scale sequencing studies.
已开发出计算机工具来预测可能影响前体mRNA剪接的变异。将这些工具应用于基础研究和临床实践的主要限制在于难以解读其输出结果。大多数工具仅根据DNA序列预测潜在的剪接位点,而不测量变异导致的剪接信号变化。另一个限制是缺乏对这些工具的大规模评估研究。我们使用受试者工作特征分析,在剪接共有区域内的2959个单核苷酸变异(scSNV)上比较了八种计算机工具。位置权重矩阵模型和最大熵扫描的表现优于其他方法。两种集成学习方法,即自适应增强和随机森林,被用于构建利用个体方法优势的模型。这两种模型都进一步提高了预测能力,其输出为可直接解读的预测分数。我们将我们的集成分数应用于来自癌症体细胞突变目录数据库的scSNV。分析表明,预测的剪接改变scSNV在复发性scSNV和已知癌症基因中富集。我们预先计算了全人类基因组中所有潜在scSNV的集成分数,为识别从大规模测序研究中发现的剪接改变scSNV提供了一个全基因组水平的资源。