Abo Ryan P, Ducar Matthew, Garcia Elizabeth P, Thorner Aaron R, Rojas-Rudilla Vanesa, Lin Ling, Sholl Lynette M, Hahn William C, Meyerson Matthew, Lindeman Neal I, Van Hummelen Paul, MacConaill Laura E
Center for Cancer Genome Discovery and Department of Medical Oncology, Dana-Farber Cancer Institute and Harvard Medical School, Boston, MA 02215, USA.
Department of Pathology, Brigham and Women's Hospital, Boston, MA 02215, USA.
Nucleic Acids Res. 2015 Feb 18;43(3):e19. doi: 10.1093/nar/gku1211. Epub 2014 Nov 26.
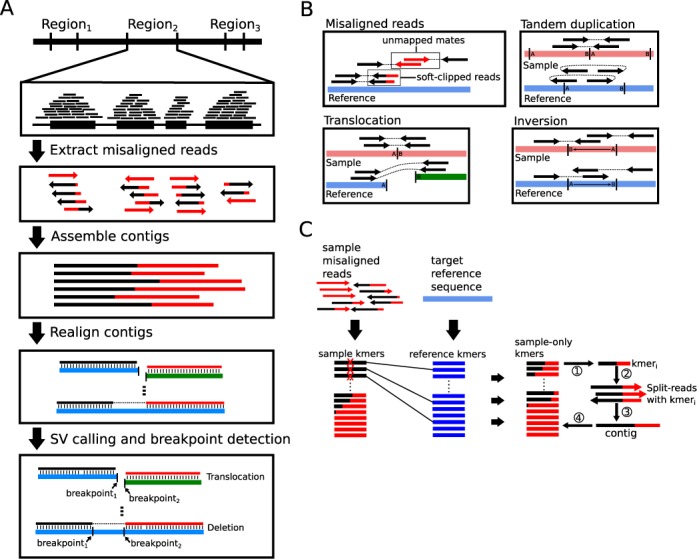
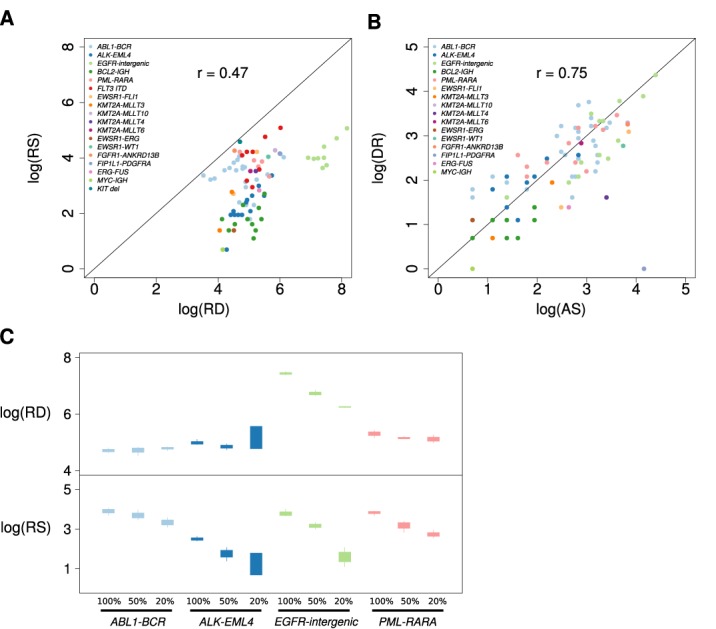
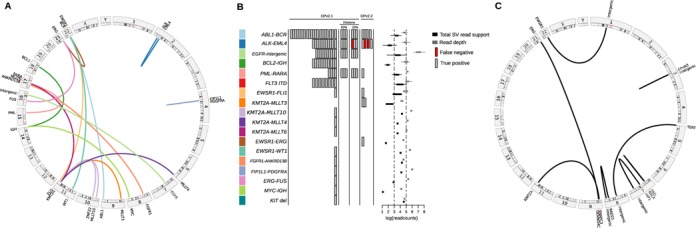
Genomic structural variation (SV), a common hallmark of cancer, has important predictive and therapeutic implications. However, accurately detecting SV using high-throughput sequencing data remains challenging, especially for 'targeted' resequencing efforts. This is critically important in the clinical setting where targeted resequencing is frequently being applied to rapidly assess clinically actionable mutations in tumor biopsies in a cost-effective manner. We present BreaKmer, a novel approach that uses a 'kmer' strategy to assemble misaligned sequence reads for predicting insertions, deletions, inversions, tandem duplications and translocations at base-pair resolution in targeted resequencing data. Variants are predicted by realigning an assembled consensus sequence created from sequence reads that were abnormally aligned to the reference genome. Using targeted resequencing data from tumor specimens with orthogonally validated SV, non-tumor samples and whole-genome sequencing data, BreaKmer had a 97.4% overall sensitivity for known events and predicted 17 positively validated, novel variants. Relative to four publically available algorithms, BreaKmer detected SV with increased sensitivity and limited calls in non-tumor samples, key features for variant analysis of tumor specimens in both the clinical and research settings.
基因组结构变异(SV)是癌症的一个常见特征,具有重要的预测和治疗意义。然而,利用高通量测序数据准确检测SV仍然具有挑战性,尤其是对于“靶向”重测序工作而言。这在临床环境中至关重要,因为靶向重测序经常被用于以具有成本效益的方式快速评估肿瘤活检中具有临床可操作性的突变。我们提出了BreaKmer,这是一种新颖的方法,它使用“kmer”策略来组装比对错误的序列读数,以在靶向重测序数据中以碱基对分辨率预测插入、缺失、倒位、串联重复和易位。通过重新比对由与参考基因组异常比对的序列读数创建的组装一致序列来预测变异。使用来自具有经正交验证的SV的肿瘤标本、非肿瘤样本的靶向重测序数据和全基因组测序数据,BreaKmer对已知事件的总体灵敏度为97.4%,并预测了17个经阳性验证的新变异。相对于四种公开可用的算法,BreaKmer检测SV的灵敏度更高,且在非肿瘤样本中的假阳性调用有限,这是临床和研究环境中肿瘤标本变异分析的关键特征。