Starenki D, Hong S-K, Lloyd R V, Park J-I
Department of Biochemistry, Medical College of Wisconsin, Milwaukee, WI, USA.
Department of Pathology and Laboratory Medicine, University of Wisconsin, Madison, WI, USA.
Oncogene. 2015 Aug 27;34(35):4624-34. doi: 10.1038/onc.2014.392. Epub 2014 Dec 1.
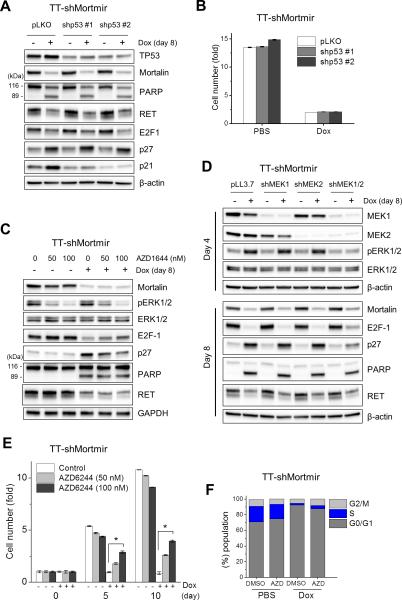
Medullary thyroid carcinoma (MTC) is a neuroendocrine tumor mainly caused by mutations in the rearranged during transfection (RET) proto-oncogene. For therapy of advanced MTC, the Food and Drug Administration recently approved vandetanib and cabozantinib, the tyrosine kinase inhibitors targeting RET, vascular endothelial growth factor receptor, epidermal growth factor receptor and/or c-MET. Nevertheless, not all patients respond to these drugs, demanding additional therapeutic strategies. We found that mortalin (HSPA9/GRP75), a member of HSP70 family, is upregulated in human MTC tissues and that its depletion robustly induces cell death and growth arrest in MTC cell lines in culture and in mouse xenografts. These effects were accompanied by substantial downregulation of RET, induction of the tumor-suppressor TP53 and altered expression of cell cycle regulatory machinery and apoptosis markers, including E2F-1, p21(CIP1), p27(KIP1) and Bcl-2 family proteins. Our investigation of the molecular mechanisms underlying these effects revealed that mortalin depletion induces transient MEK/ERK (extracellular signal-regulated kinase) activation and altered mitochondrial bioenergetics in MTC cells, as indicated by depolarized mitochondrial membrane, decreased oxygen consumption and extracellular acidification and increased oxidative stress. Intriguingly, mortalin depletion induced growth arrest partly via the MEK/ERK pathway, whereas it induced cell death by causing mitochondrial dysfunction in a Bcl-2-dependent manner. However, TP53 was not necessary for these effects except for p21(CIP1) induction. Moreover, mortalin depletion downregulated RET expression independently of MEK/ERK and TP53. These data demonstrate that mortalin is a key regulator of multiple signaling and metabolic pathways pivotal to MTC cell survival and proliferation, proposing mortalin as a novel therapeutic target for MTC.
甲状腺髓样癌(MTC)是一种神经内分泌肿瘤,主要由转染期间重排(RET)原癌基因突变引起。对于晚期MTC的治疗,美国食品药品监督管理局最近批准了凡德他尼和卡博替尼,这两种酪氨酸激酶抑制剂可靶向RET、血管内皮生长因子受体、表皮生长因子受体和/或c-MET。然而,并非所有患者对这些药物都有反应,因此需要额外的治疗策略。我们发现,热休克蛋白70(HSP70)家族成员mortalin(HSPA9/GRP75)在人MTC组织中上调,其缺失可在培养的MTC细胞系和小鼠异种移植模型中强烈诱导细胞死亡和生长停滞。这些效应伴随着RET的显著下调、肿瘤抑制因子TP53的诱导以及细胞周期调节机制和凋亡标志物(包括E2F-1、p21(CIP1)、p27(KIP1)和Bcl-2家族蛋白)表达的改变。我们对这些效应背后分子机制的研究表明,mortalin缺失可诱导MTC细胞中MEK/ERK(细胞外信号调节激酶)的短暂激活和线粒体生物能量学的改变,表现为线粒体膜去极化、氧消耗减少、细胞外酸化增加以及氧化应激增加。有趣的是,mortalin缺失部分通过MEK/ERK途径诱导生长停滞,而通过以Bcl-2依赖的方式导致线粒体功能障碍诱导细胞死亡。然而,除了诱导p21(CIP1)外,这些效应并不需要TP53。此外,mortalin缺失独立于MEK/ERK和TP53下调RET表达。这些数据表明,mortalin是MTC细胞存活和增殖关键的多种信号和代谢途径的关键调节因子,提示mortalin可作为MTC的新型治疗靶点。