Kurotani Atsushi, Yamada Yutaka, Shinozaki Kazuo, Kuroda Yutaka, Sakurai Tetsuya
RIKEN Center for Sustainable Resource Science, Yokohama, Kanagawa, 230-0045 Japan Department of Biotechnology and Life Sciences, Faculty of Technology, Tokyo University of Agriculture and Technology, Koganei, Tokyo, 184-8588 Japan.
RIKEN Center for Sustainable Resource Science, Yokohama, Kanagawa, 230-0045 Japan.
Plant Cell Physiol. 2015 Jan;56(1):e11. doi: 10.1093/pcp/pcu176. Epub 2014 Nov 29.
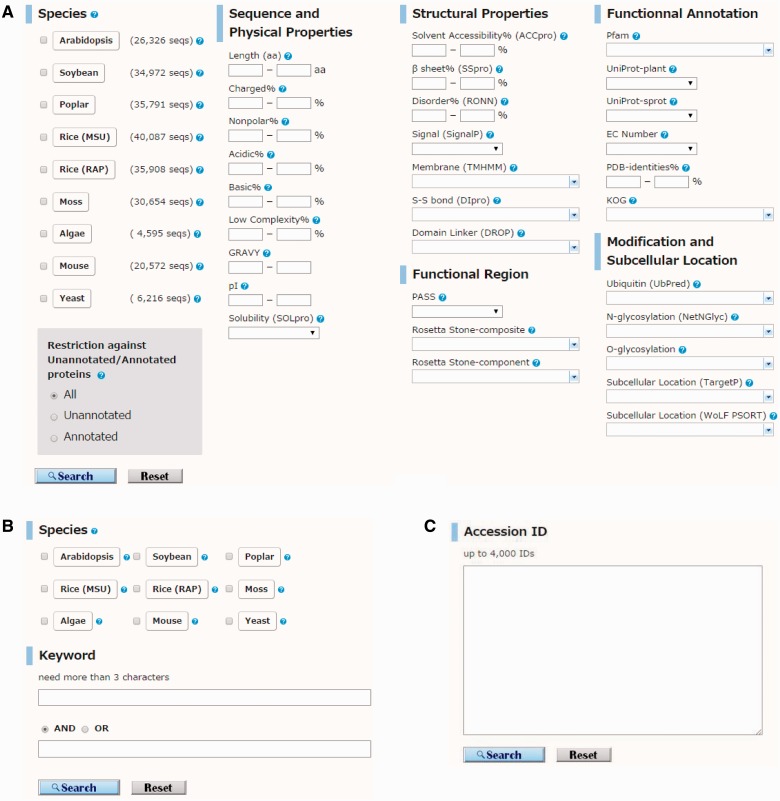
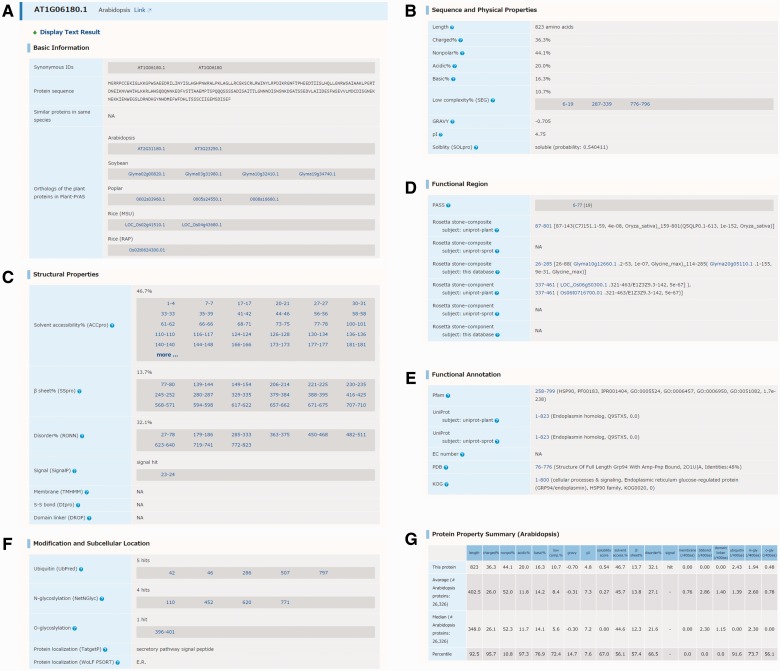
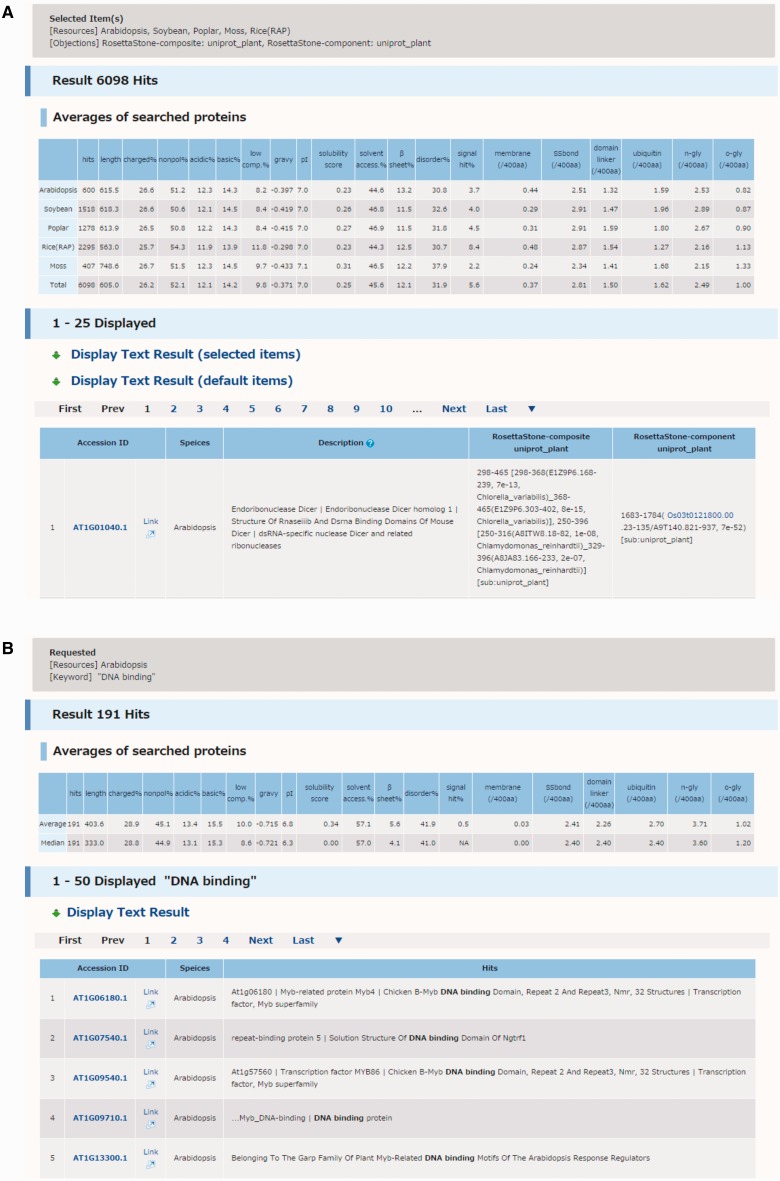
Arabidopsis thaliana is an important model species for studies of plant gene functions. Research on Arabidopsis has resulted in the generation of high-quality genome sequences, annotations and related post-genomic studies. The amount of annotation, such as gene-coding regions and structures, is steadily growing in the field of plant research. In contrast to the genomics resource of animals and microorganisms, there are still some difficulties with characterization of some gene functions in plant genomics studies. The acquisition of information on protein structure can help elucidate the corresponding gene function because proteins encoded in the genome possess highly specific structures and functions. In this study, we calculated multiple physicochemical and secondary structural parameters of protein sequences, including length, hydrophobicity, the amount of secondary structure, the number of intrinsically disordered regions (IDRs) and the predicted presence of transmembrane helices and signal peptides, using a total of 208,333 protein sequences from the genomes of six representative plant species, Arabidopsis thaliana, Glycine max (soybean), Populus trichocarpa (poplar), Oryza sativa (rice), Physcomitrella patens (moss) and Cyanidioschyzon merolae (alga). Using the PASS tool and the Rosetta Stone method, we annotated the presence of novel functional regions in 1,732 protein sequences that included unannotated sequences from the Arabidopsis and rice proteomes. These results were organized into the Plant Protein Annotation Suite database (Plant-PrAS), which can be freely accessed online at http://plant-pras.riken.jp/.
拟南芥是用于植物基因功能研究的重要模式物种。对拟南芥的研究已产生了高质量的基因组序列、注释及相关的后基因组研究成果。在植物研究领域,诸如基因编码区和结构等注释信息的数量正在稳步增长。与动物和微生物的基因组学资源相比,植物基因组学研究中某些基因功能的表征仍存在一些困难。获取蛋白质结构信息有助于阐明相应的基因功能,因为基因组中编码的蛋白质具有高度特异性的结构和功能。在本研究中,我们使用来自六种代表性植物物种(拟南芥、大豆、毛果杨、水稻、小立碗藓和聚球藻)基因组的总共208,333个蛋白质序列,计算了蛋白质序列的多个物理化学和二级结构参数,包括长度、疏水性、二级结构数量、内在无序区域(IDR)数量以及跨膜螺旋和信号肽的预测存在情况。使用PASS工具和罗塞塔石碑方法,我们注释了1732个蛋白质序列中新型功能区域的存在情况,这些序列包括来自拟南芥和水稻蛋白质组的未注释序列。这些结果被整理到植物蛋白质注释套件数据库(Plant-PrAS)中,该数据库可在http://plant-pras.riken.jp/在线免费访问。