Righetto Irene, Milani Adelaide, Cattoli Giovanni, Filippini Francesco
Molecular Biology and Bioinformatics Unit (MOLBINFO), Department of Biology, University of Padua, via U. Bassi 58/B, 35131, Padova, Italy.
FAO-OIE and National Reference Laboratory for Newcastle Disease and Avian Influenza, Istituto Zooprofilattico delle Venezie (IZSVe), viale dell'Università 10, 35020, Legnaro, Italy.
BMC Bioinformatics. 2014 Dec 10;15(1):363. doi: 10.1186/s12859-014-0363-5.
Genome variation is very high in influenza A viruses. However, viral evolution and spreading is strongly influenced by immunogenic features and capacity to bind host cells, depending in turn on the two major capsidic proteins. Therefore, such viruses are classified based on haemagglutinin and neuraminidase types, e.g. H5N1. Current analyses of viral evolution are based on serological and primary sequence comparison; however, comparative structural analysis of capsidic proteins can provide functional insights on surface regions possibly crucial to antigenicity and cell binding.
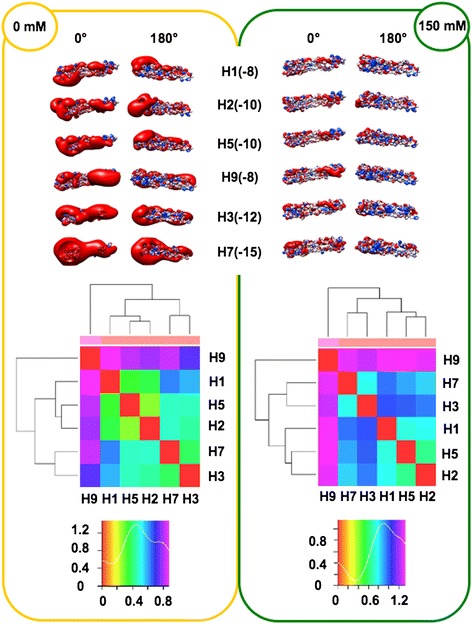
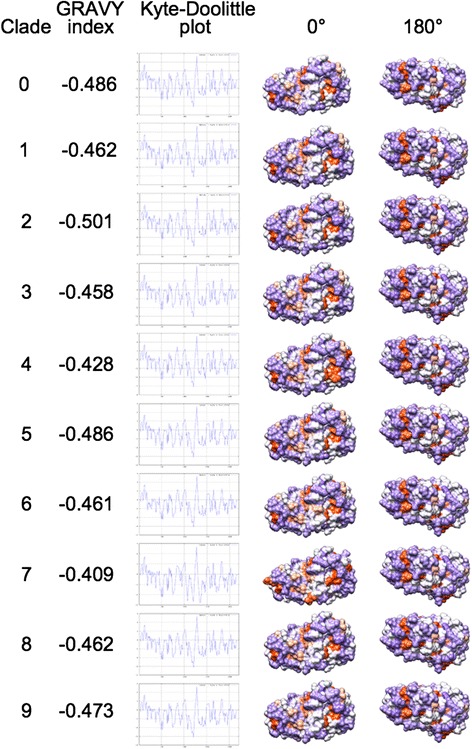
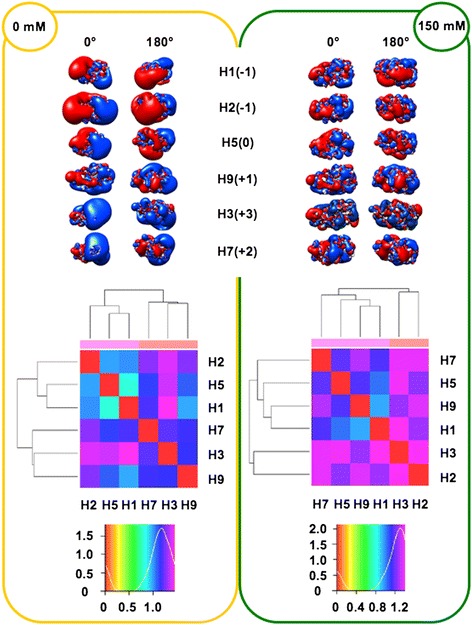
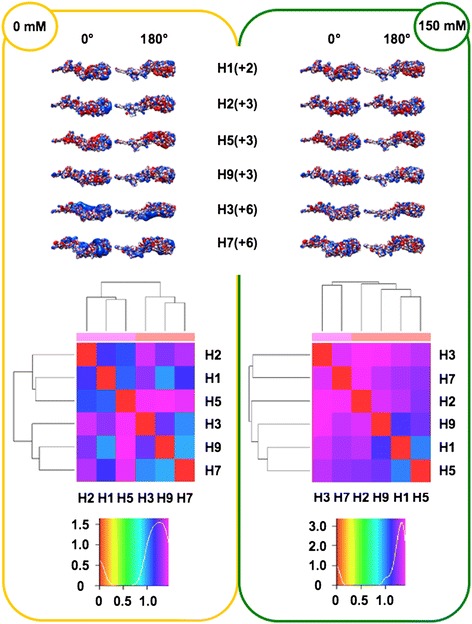
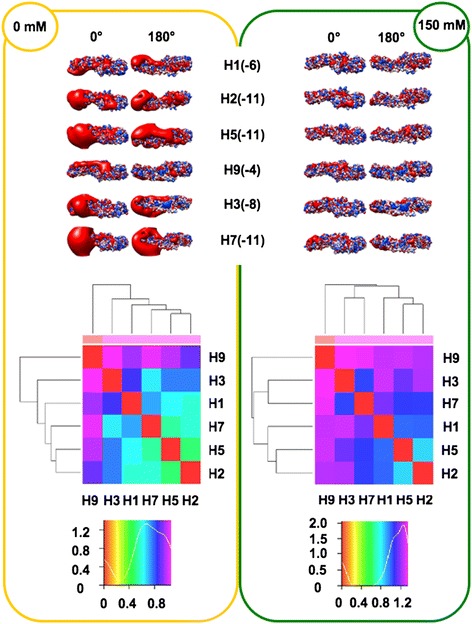
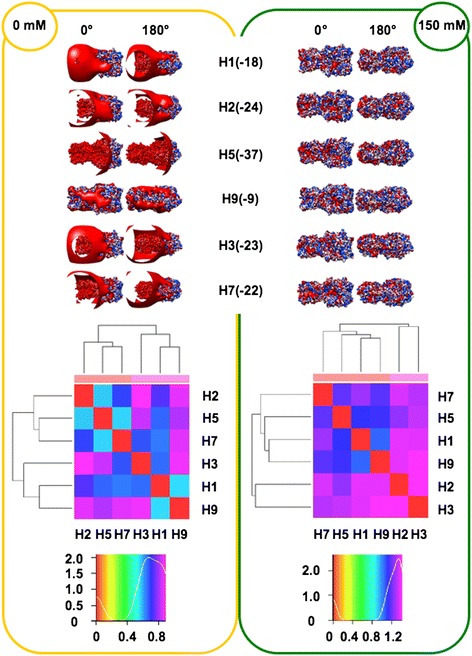
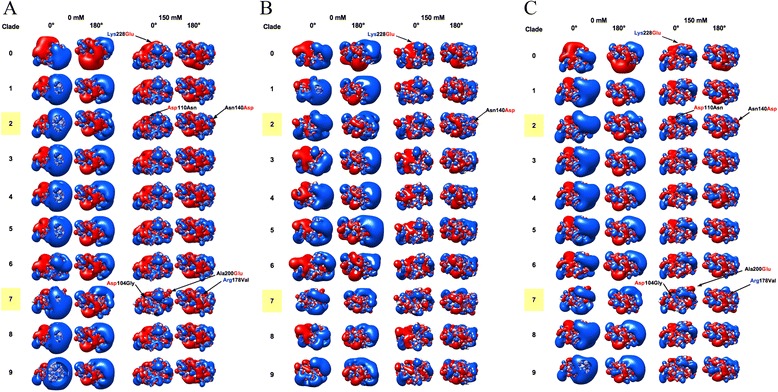
We performed extensive structural comparison of influenza virus haemagglutinins and of their domains and subregions to investigate type- and/or domain-specific variation. We found that structural closeness and primary sequence similarity are not always tightly related; moreover, type-specific features could be inferred when comparing surface properties of haemagglutinin subregions, monomers and trimers, in terms of electrostatics and hydropathy. Focusing on H5N1, we found that variation at the receptor binding domain surface intriguingly relates to branching of still circulating clades from those ones that are no longer circulating.
Evidence from this work suggests that integrating phylogenetic and serological analyses by extensive structural comparison can help in understanding the 'functional evolution' of viral surface determinants. In particular, variation in electrostatic and hydropathy patches can provide molecular evolution markers: intriguing surface charge redistribution characterizing the haemagglutinin receptor binding domains from circulating H5N1 clades 2 and 7 might have contributed to antigenic escape hence to their evolutionary success and spreading.
甲型流感病毒的基因组变异程度很高。然而,病毒的进化和传播受到免疫原性特征以及结合宿主细胞能力的强烈影响,而这又反过来取决于两种主要的衣壳蛋白。因此,此类病毒是根据血凝素和神经氨酸酶类型进行分类的,例如H5N1。目前对病毒进化的分析基于血清学和一级序列比较;然而,衣壳蛋白的比较结构分析可以为可能对抗抗原性和细胞结合至关重要的表面区域提供功能见解。
我们对流感病毒血凝素及其结构域和亚区域进行了广泛的结构比较,以研究类型和/或结构域特异性变异。我们发现结构相似性和一级序列相似性并不总是紧密相关;此外,在比较血凝素亚区域、单体和三聚体的表面性质时,从静电学和亲水性角度可以推断出类型特异性特征。聚焦于H5N1,我们发现受体结合结构域表面的变异有趣地与仍在传播的分支与不再传播的分支的分化相关。
这项工作的证据表明,通过广泛的结构比较整合系统发育分析和血清学分析有助于理解病毒表面决定簇的“功能进化”。特别是,静电和亲水区域的变异可以提供分子进化标记:来自正在传播的H5N1第2和7分支的血凝素受体结合结构域中表征抗原逃逸的有趣表面电荷重新分布可能有助于它们的进化成功和传播。