Kitamura Toshio, Inoue Daichi, Okochi-Watanabe Naoko, Kato Naoko, Komeno Yukiko, Lu Yang, Enomoto Yutaka, Doki Noriko, Uchida Tomoyuki, Kagiyama Yuki, Togami Katsuhiro, Kawabata Kimihito C, Nagase Reina, Horikawa Sayuri, Hayashi Yasutaka, Saika Makoto, Fukuyama Tomofusa, Izawa Kumi, Oki Toshihiko, Nakahara Fumio, Kitaura Jiro
Division of Cellular Therapy/Division of Stem Cell Signaling, The Institute of Medical Science, The University of Tokyo.
Proc Jpn Acad Ser B Phys Biol Sci. 2014;90(10):389-404. doi: 10.2183/pjab.90.389.
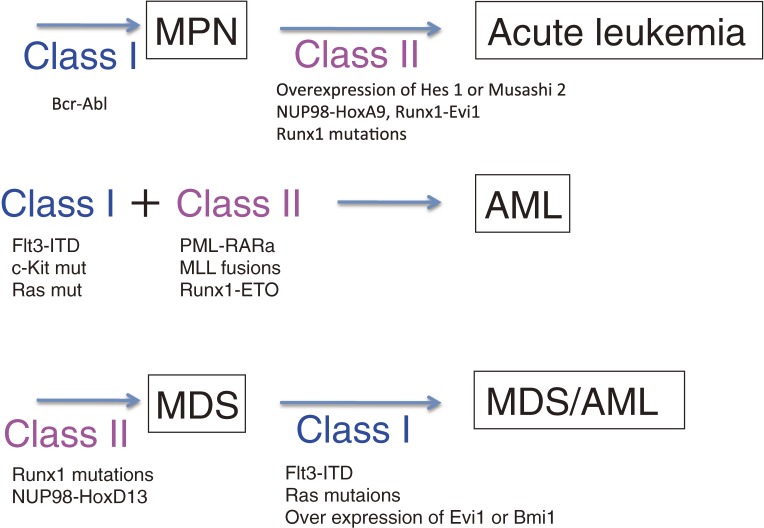
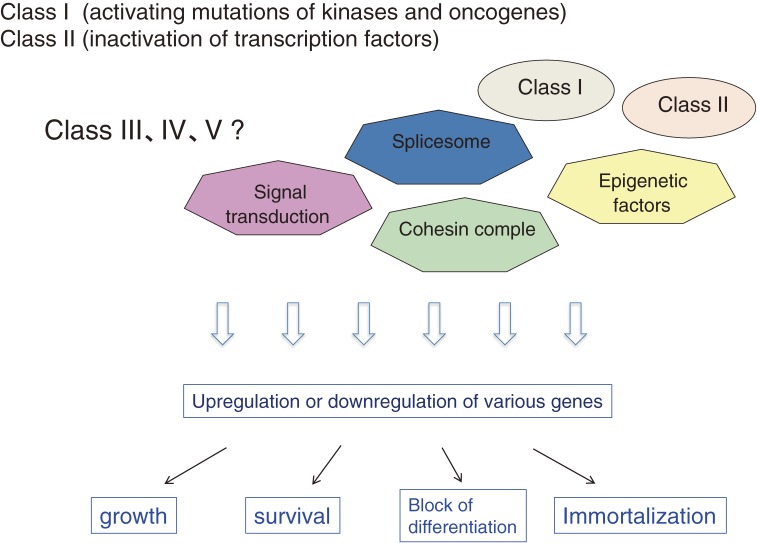
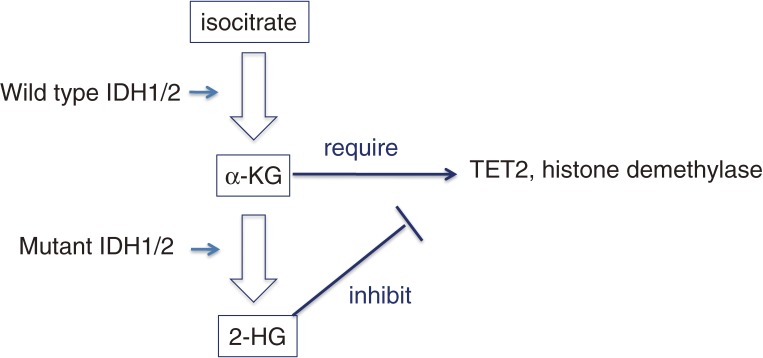
Myeloid malignancies consist of acute myeloid leukemia (AML), myelodysplastic syndromes (MDS) and myeloproliferative neoplasm (MPN). The latter two diseases have preleukemic features and frequently evolve to AML. As with solid tumors, multiple mutations are required for leukemogenesis. A decade ago, these gene alterations were subdivided into two categories: class I mutations stimulating cell growth or inhibiting apoptosis; and class II mutations that hamper differentiation of hematopoietic cells. In mouse models, class I mutations such as the Bcr-Abl fusion kinase induce MPN by themselves and some class II mutations such as Runx1 mutations induce MDS. Combinations of class I and class II mutations induce AML in a variety of mouse models. Thus, it was postulated that hematopoietic cells whose differentiation is blocked by class II mutations would autonomously proliferate with class I mutations leading to the development of leukemia. Recent progress in high-speed sequencing has enabled efficient identification of novel mutations in a variety of molecules including epigenetic factors, splicing factors, signaling molecules and proteins in the cohesin complex; most of these are not categorized as either class I or class II mutations. The functional consequences of these mutations are now being extensively investigated. In this article, we will review the molecular basis of hematological malignancies, focusing on mouse models and the interfaces between these models and clinical findings, and revisit the classical class I/II hypothesis.
髓系恶性肿瘤包括急性髓系白血病(AML)、骨髓增生异常综合征(MDS)和骨髓增殖性肿瘤(MPN)。后两种疾病具有白血病前期特征,并经常演变为AML。与实体瘤一样,白血病发生需要多个突变。十年前,这些基因改变被分为两类:I类突变刺激细胞生长或抑制细胞凋亡;II类突变阻碍造血细胞分化。在小鼠模型中,诸如Bcr-Abl融合激酶之类的I类突变自身可诱导MPN,而诸如Runx1突变之类的一些II类突变可诱导MDS。I类和II类突变的组合在多种小鼠模型中可诱导AML。因此,有人推测,其分化被II类突变阻断的造血细胞会因I类突变而自主增殖,从而导致白血病的发生。高速测序技术的最新进展已能够有效鉴定包括表观遗传因子、剪接因子、信号分子和黏连蛋白复合体中的蛋白质在内的多种分子中的新突变;其中大多数不属于I类或II类突变。目前正在广泛研究这些突变的功能后果。在本文中,我们将回顾血液系统恶性肿瘤的分子基础,重点关注小鼠模型以及这些模型与临床发现之间的联系,并重新审视经典的I/II类假说。