Ahmed Lucky, Rhaman Md Mhahabubur, Mendy John S, Wang Jing, Fronczek Frank R, Powell Douglas R, Leszczynski Jerzy, Hossain Md Alamgir
Department of Chemistry and Biochemistry, Jackson State University , Jackson, Mississippi 39217, United States.
J Phys Chem A. 2015 Jan 15;119(2):383-94. doi: 10.1021/jp511040p. Epub 2015 Jan 6.
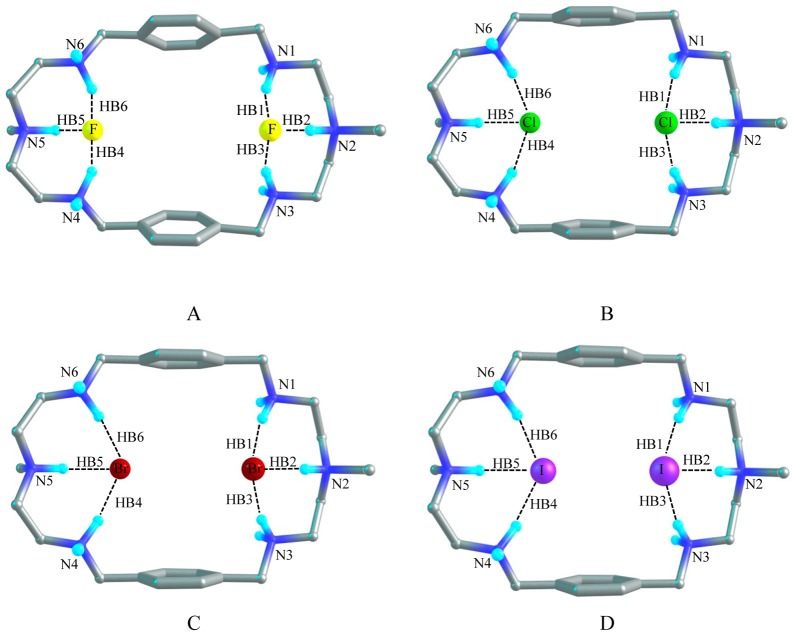
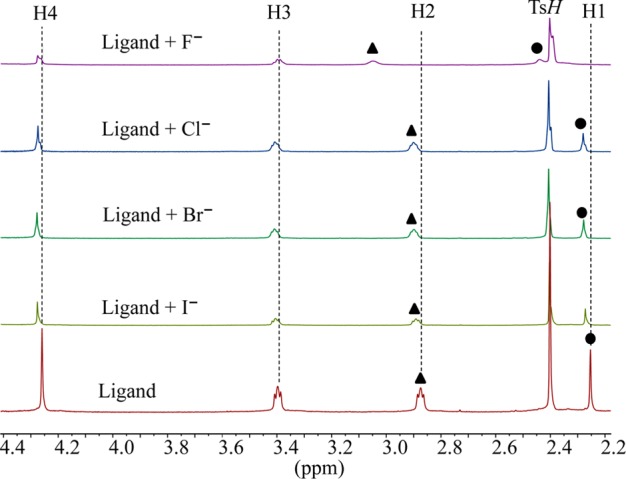
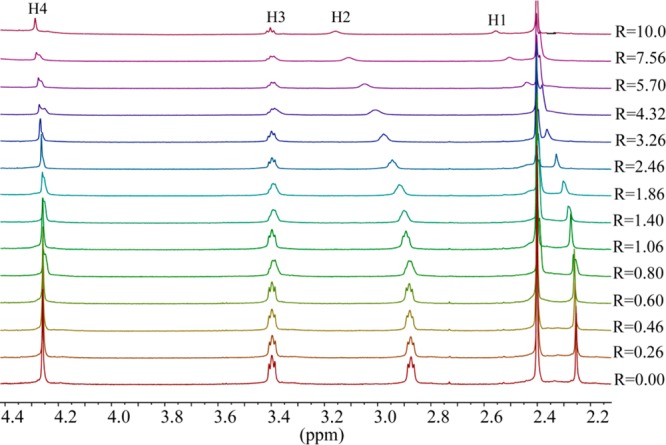
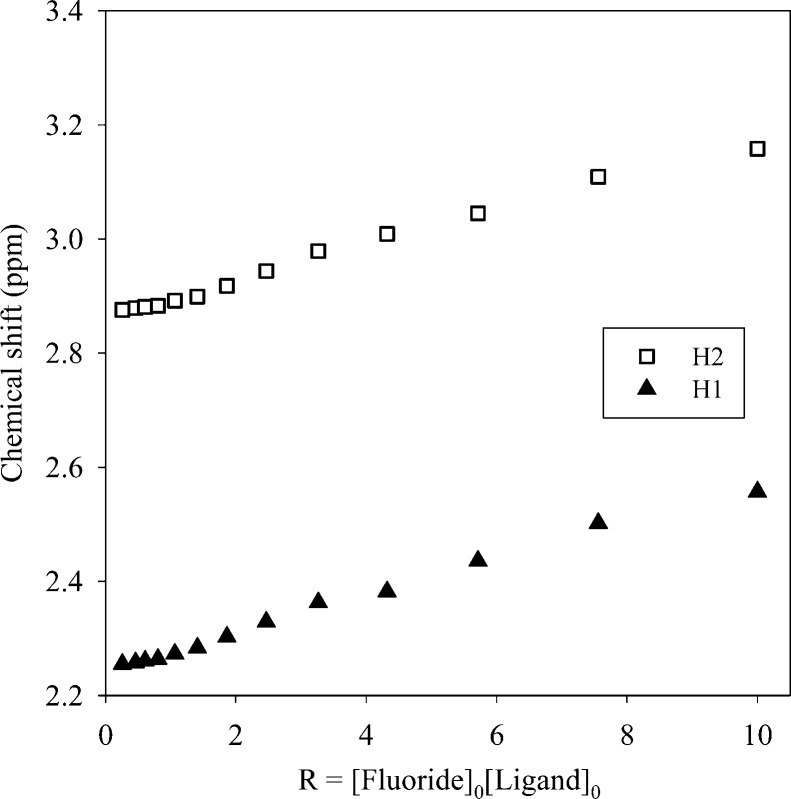
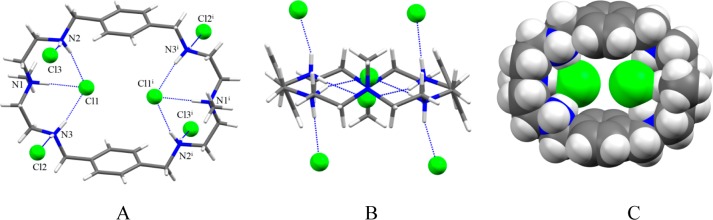
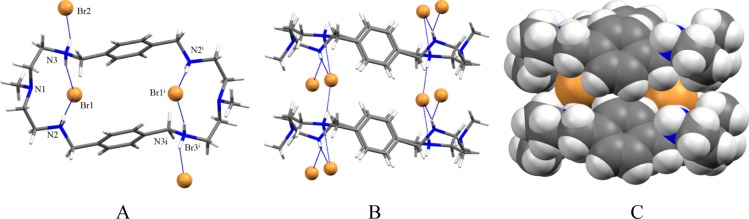
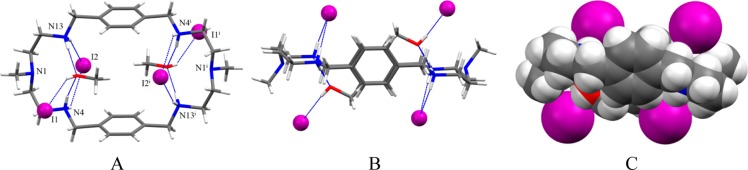
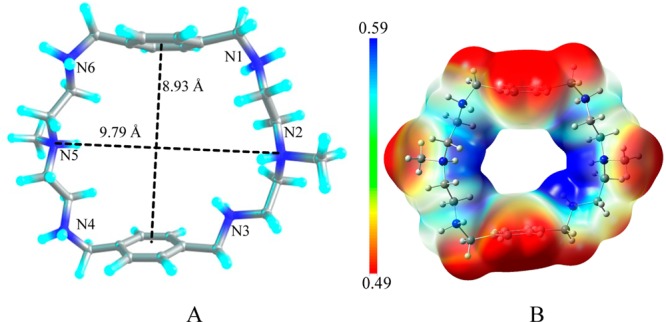
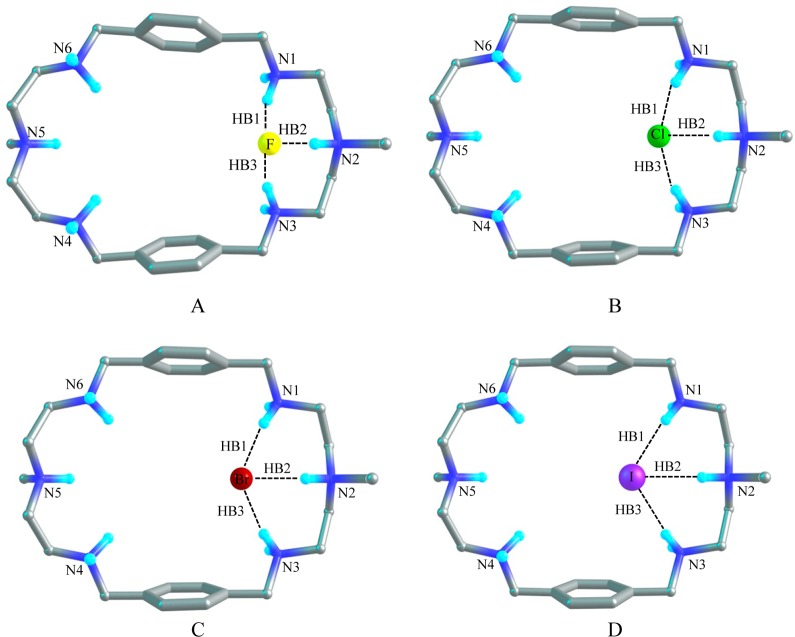
A p-xylyl-based macrocycle L has been synthesized and its binding properties with halides have been investigated by (1)H NMR titrations, single crystal X-ray diffraction analysis, and density functional theory (DFT) calculations. As investigated by (1)H NMR titrations, the ligand preferentially binds a halide in a 1:2 binding mode, with the association constants (in log K2) of 2.82, 2.70, 2.28, and 2.20 for fluoride, chloride, bromide, and iodide, respectively. The overall binding trend was found to be in the order of fluoride > chloride > bromide > iodide, reflecting that the binding strength correlates with the relative basicity and size of the respective halide. Crystallographic studies indicate that the ligand forms 1:2 complexes with chloride, bromide and iodide. In the chloride complex, the ligand is hexaprotonated and each chloride is held via three NH···Cl(-) bonds. The ligand is tetraprotonated for the other complexes, where each halide is H-bonded to two secondary ammonium NH(+) groups via NH···X(-) bonds. The results of DFT calculations performed on H6L at M062x/6-311G (d,p) level in both gas and solvent phases, suggest that the ligand binds halides with the binding energy in the order of F(-) > Cl(-) > Br(-) > I(-), supporting the experimental data obtained from (1)H NMR studies. Results from DFT calculations further indicate that a 1:2 binding is energetically more favorable than a 1:1 binding of the ligand.
已合成了一种对二甲苯基大环化合物L,并通过¹H NMR滴定、单晶X射线衍射分析和密度泛函理论(DFT)计算研究了其与卤化物的结合性质。通过¹H NMR滴定研究发现,该配体以1:2的结合模式优先结合卤化物,氟化物、氯化物、溴化物和碘化物的缔合常数(以log K₂计)分别为2.82、2.70、2.28和2.20。发现整体结合趋势为氟化物>氯化物>溴化物>碘化物,这反映出结合强度与相应卤化物的相对碱性和大小相关。晶体学研究表明,该配体与氯化物、溴化物和碘化物形成1:2配合物。在氯化物配合物中,配体为六质子化,每个氯化物通过三个NH···Cl⁻键固定。对于其他配合物,配体为四质子化,其中每个卤化物通过NH···X⁻键与两个仲铵NH⁺基团形成氢键。在气相和溶剂相中,在M062x/6 - 311G(d,p)水平上对[H₆L]⁶⁺进行DFT计算的结果表明,配体与卤化物的结合能顺序为F⁻>Cl⁻>Br⁻>I⁻,支持了从¹H NMR研究中获得的实验数据。DFT计算结果进一步表明,配体以1:2结合在能量上比1:1结合更有利。