Carlson Brian M, Onusko Samuel W, Gross Joshua B
Department of Biological Sciences, University of Cincinnati, Cincinnati, Ohio 45221.
Department of Biological Sciences, University of Cincinnati, Cincinnati, Ohio 45221
G3 (Bethesda). 2014 Dec 17;5(2):241-51. doi: 10.1534/g3.114.015438.
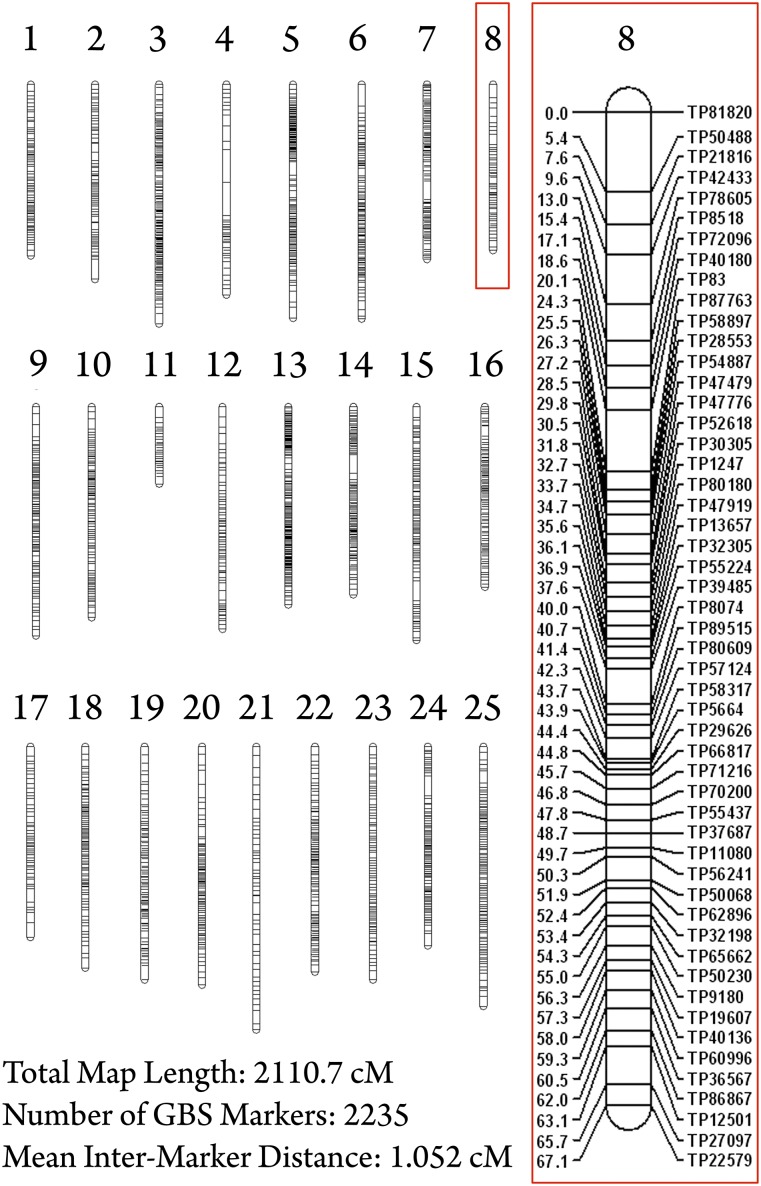
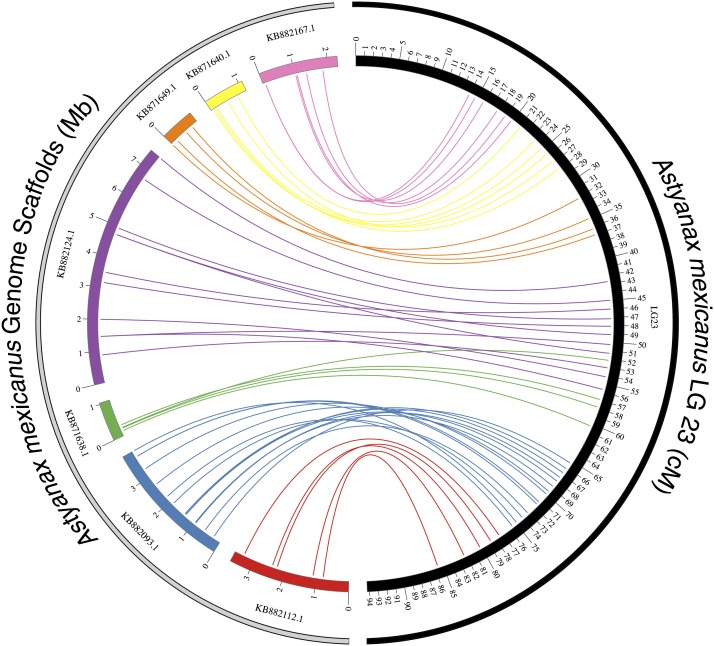
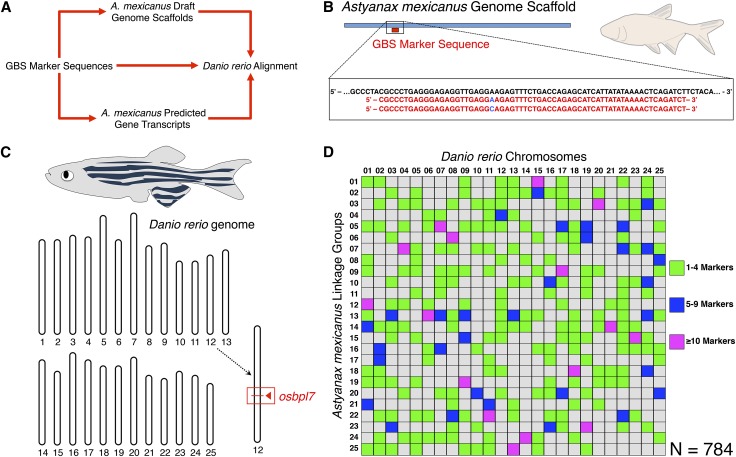
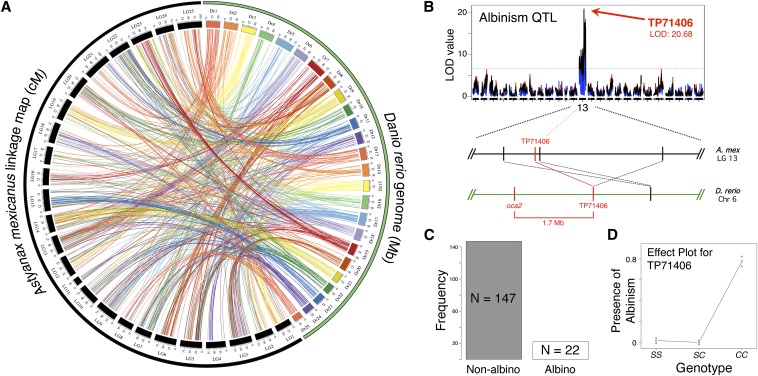
The Mexican tetra, Astyanax mexicanus, is a unique model system consisting of cave-adapted and surface-dwelling morphotypes that diverged >1 million years (My) ago. This remarkable natural experiment has enabled powerful genetic analyses of cave adaptation. Here, we describe the application of next-generation sequencing technology to the creation of a high-density linkage map. Our map comprises more than 2200 markers populating 25 linkage groups constructed from genotypic data generated from a single genotyping-by-sequencing project. We leveraged emergent genomic and transcriptomic resources to anchor hundreds of anonymous Astyanax markers to the genome of the zebrafish (Danio rerio), the most closely related model organism to our study species. This facilitated the identification of 784 distinct connections between our linkage map and the Danio rerio genome, highlighting several regions of conserved genomic architecture between the two species despite ~150 My of divergence. Using a Mendelian cave-associated trait as a proof-of-principle, we successfully recovered the genomic position of the albinism locus near the gene Oca2. Further, our map successfully informed the positions of unplaced Astyanax genomic scaffolds within particular linkage groups. This ability to identify the relative location, orientation, and linear order of unaligned genomic scaffolds will facilitate ongoing efforts to improve on the current early draft and assemble future versions of the Astyanax physical genome. Moreover, this improved linkage map will enable higher-resolution genetic analyses and catalyze the discovery of the genetic basis for cave-associated phenotypes.
墨西哥丽脂鲤(Astyanax mexicanus)是一种独特的模型系统,由洞穴适应型和地表栖息型形态组成,它们在100多万年前就已经分化。这个非凡的自然实验使得对洞穴适应进行强大的基因分析成为可能。在这里,我们描述了下一代测序技术在创建高密度连锁图谱中的应用。我们的图谱包含2200多个标记,分布在25个连锁群中,这些连锁群是根据一个单一的测序分型项目生成的基因型数据构建的。我们利用新兴的基因组和转录组资源,将数百个匿名的丽脂鲤标记定位到斑马鱼(Danio rerio)的基因组上,斑马鱼是与我们研究物种关系最密切的模式生物。这有助于识别我们的连锁图谱与斑马鱼基因组之间的784个不同的联系,突出了两个物种之间尽管有大约1.5亿年的分化,但仍存在几个保守的基因组结构区域。以孟德尔式的洞穴相关性状作为原理验证,我们成功地找到了白化病基因座在Oca2基因附近的基因组位置。此外,我们的图谱成功地确定了未定位的丽脂鲤基因组支架在特定连锁群中的位置。这种识别未对齐基因组支架的相对位置、方向和线性顺序的能力,将有助于当前改进早期草稿和组装丽脂鲤物理基因组未来版本的工作。此外,这个改进的连锁图谱将能够进行更高分辨率的基因分析,并促进发现洞穴相关表型的遗传基础。