Chen Kuan-Bei, Hardison Ross, Zhang Yu
BMC Genomics. 2014;15 Suppl 9(Suppl 9):S12. doi: 10.1186/1471-2164-15-S9-S12. Epub 2014 Dec 8.
Current ChIP-seq studies are interested in comparing multiple epigenetic profiles across several cell types and tissues simultaneously for studying constitutive and differential regulation. Simultaneous analysis of multiple epigenetic features in many samples can gain substantial power and specificity than analyzing individual features and/or samples separately. Yet there are currently few tools can perform joint inference of constitutive and differential regulation in multi-feature-multi-condition contexts with statistical testing. Existing tools either test regulatory variation for one factor in multiple samples at a time, or for multiple factors in one or two samples. Many of them only identify binary rather than quantitative variation, which are sensitive to threshold choices.
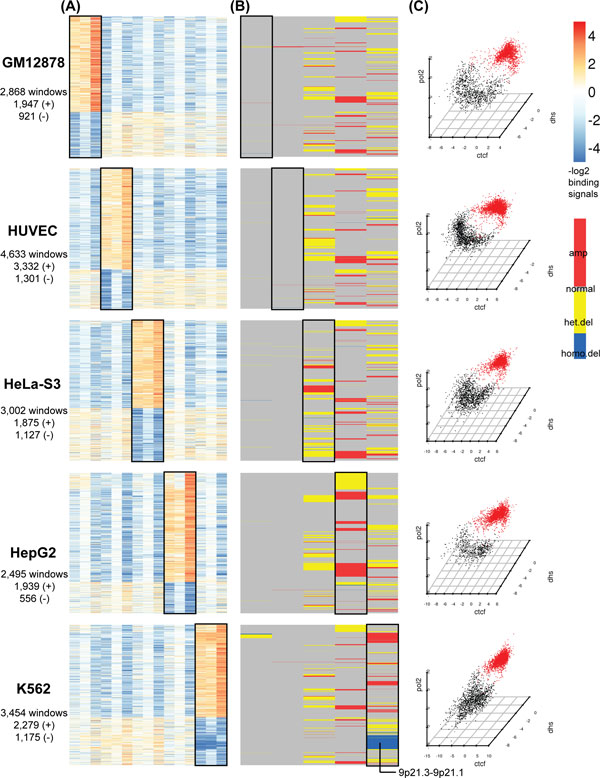
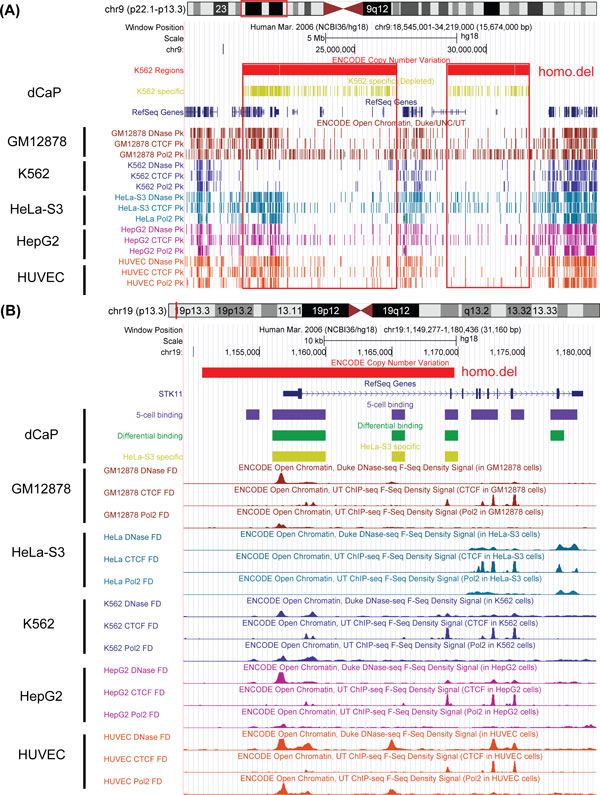
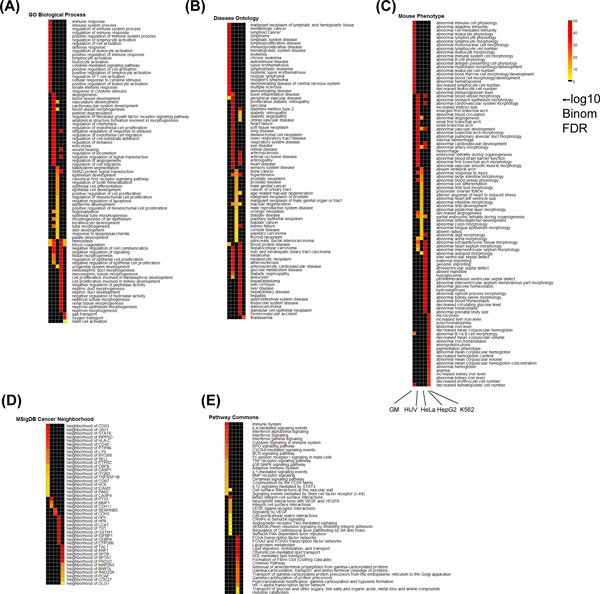
We propose a novel and powerful method called dCaP for simultaneously detecting constitutive and differential regulation of multiple epigenetic factors in multiple samples. Using simulation, we demonstrate the superior power of dCaP compared to existing methods. We then apply dCaP to two datasets from human and mouse ENCODE projects to demonstrate its utility. We show in the human dataset that the cell-type specific regulatory loci detected by dCaP are significantly enriched near genes with cell-type specific functions and disease relevance. We further show in the mouse dataset that dCaP captures genomic regions showing significant signal variations for TAL1 occupancy between two mouse erythroid cell lines. The novel TAL1 occupancy loci detected only by dCaP are highly enriched with GATA1 occupancy and differential gene expression, while those detected only by other methods are not.
Here, we developed a novel approach to utilize the cooperative property of proteins to detect differential binding given multivariate ChIP-seq samples to provide better power, aiming for complementing existing approaches and providing new insights in the method development in this field.
当前的染色质免疫沉淀测序(ChIP-seq)研究旨在同时比较多种细胞类型和组织中的多个表观遗传图谱,以研究组成型和差异调控。与单独分析单个特征和/或样本相比,同时分析多个样本中的多种表观遗传特征可以获得更强的功效和特异性。然而,目前很少有工具能够在多特征-多条件背景下通过统计检验对组成型和差异调控进行联合推断。现有的工具要么一次测试多个样本中一个因素的调控变异,要么测试一两个样本中多个因素的调控变异。其中许多工具只能识别二元而非定量变异,这对阈值选择很敏感。
我们提出了一种名为dCaP的新颖且强大的方法,用于同时检测多个样本中多个表观遗传因子的组成型和差异调控。通过模拟,我们证明了dCaP与现有方法相比具有更高的功效。然后,我们将dCaP应用于来自人类和小鼠ENCODE项目的两个数据集,以证明其效用。我们在人类数据集中表明,dCaP检测到的细胞类型特异性调控位点在具有细胞类型特异性功能和疾病相关性的基因附近显著富集。我们在小鼠数据集中进一步表明,dCaP捕获了在两种小鼠红细胞系之间显示TAL1占据显著信号变化的基因组区域。仅由dCaP检测到的新型TAL1占据位点高度富集GATA1占据和差异基因表达,而仅由其他方法检测到的位点则不然。
在此,我们开发了一种新颖的方法,利用蛋白质的协同特性,在多变量ChIP-seq样本中检测差异结合,以提供更好的功效,旨在补充现有方法并为该领域的方法开发提供新的见解。