Kou Qiang, Wu Si, Liu Xiaowen
Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, 535 W, Michigan Street, Indianapolis, IN 46202, USA.
BMC Genomics. 2014 Dec 18;15(1):1140. doi: 10.1186/1471-2164-15-1140.
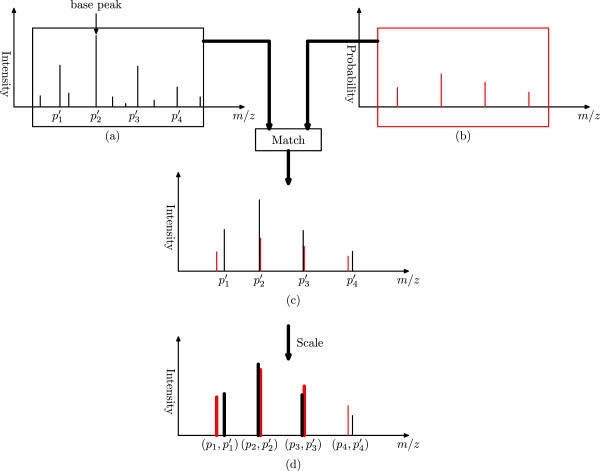
Top-down mass spectrometry plays an important role in intact protein identification and characterization. Top-down mass spectra are more complex than bottom-up mass spectra because they often contain many isotopomer envelopes from highly charged ions, which may overlap with one another. As a result, spectral deconvolution, which converts a complex top-down mass spectrum into a monoisotopic mass list, is a key step in top-down spectral interpretation.
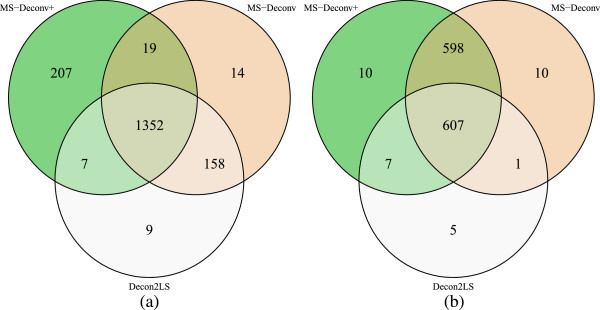
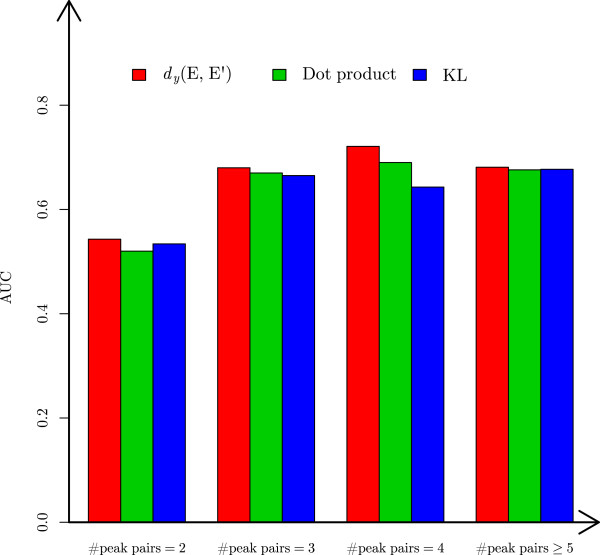
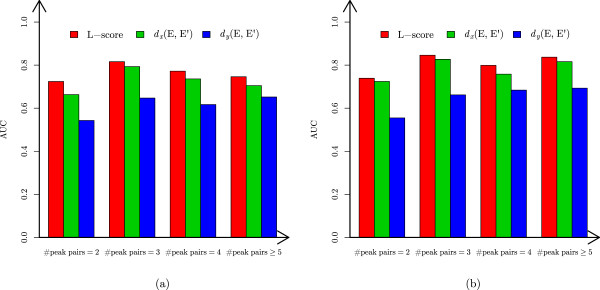
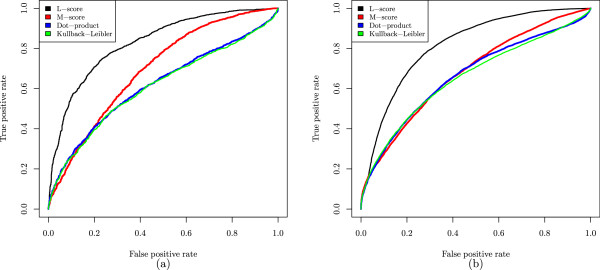
In this paper, we propose a new scoring function, L-score, for evaluating isotopomer envelopes. By combining L-score with MS-Deconv, a new software tool, MS-Deconv+, was developed for top-down spectral deconvolution. Experimental results showed that MS-Deconv+ outperformed existing software tools in top-down spectral deconvolution.
L-score shows high discriminative ability in identification of isotopomer envelopes. Using L-score, MS-Deconv+ reports many correct monoisotopic masses missed by other software tools, which are valuable for proteoform identification and characterization.
自上而下的质谱分析在完整蛋白质的鉴定和表征中起着重要作用。自上而下的质谱图比自下而上的质谱图更复杂,因为它们通常包含许多来自高电荷离子的同位素异构体包络,这些包络可能相互重叠。因此,将复杂的自上而下质谱图转换为单同位素质量列表的谱图去卷积是自上而下谱图解释的关键步骤。
在本文中,我们提出了一种用于评估同位素异构体包络的新评分函数L-score。通过将L-score与新软件工具MS-Deconv相结合,开发了用于自上而下谱图去卷积的新软件工具MS-Deconv+。实验结果表明,MS-Deconv+在自上而下谱图去卷积方面优于现有软件工具。
L-score在同位素异构体包络的识别中显示出高判别能力。使用L-score,MS-Deconv+报告了许多其他软件工具遗漏的正确单同位素质量,这对蛋白质异构体的鉴定和表征很有价值。