Cornejo Omar E, Fisher David, Escalante Ananias A
School of Biological Sciences, Washington State University
Center for Evolutionary Medicine and Informatics, the Biodesign Institute, Arizona State University.
Genome Biol Evol. 2014 Dec 17;7(1):106-19. doi: 10.1093/gbe/evu267.
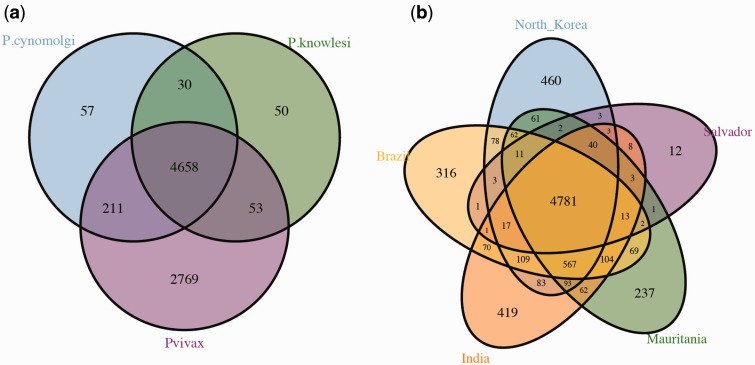
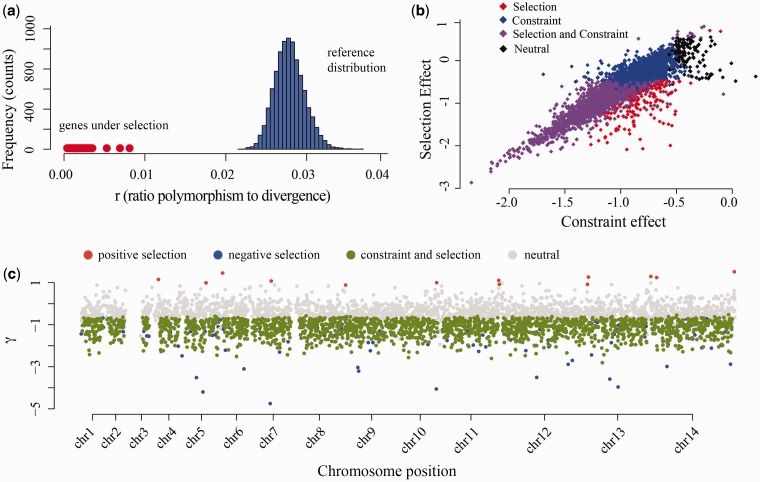
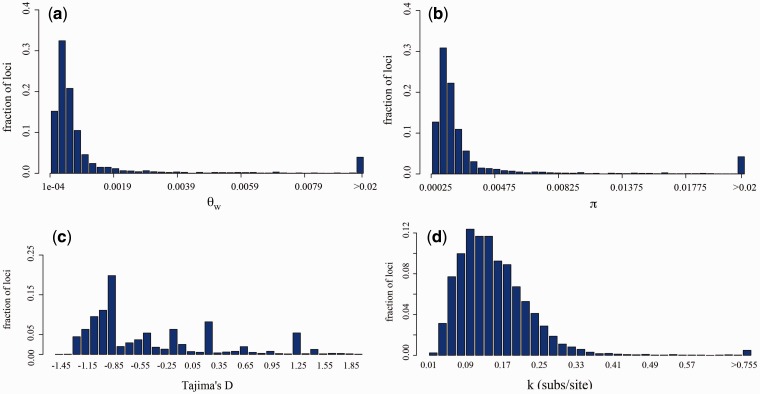
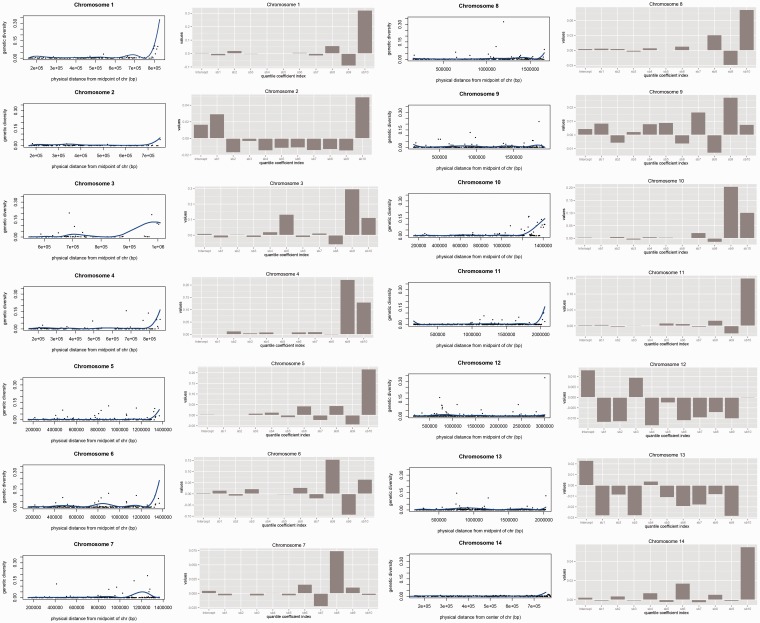
Plasmodium vivax is the most prevalent human malaria parasite outside of Africa. Yet, studies aimed to identify genes with signatures consistent with natural selection are rare. Here, we present a comparative analysis of the pattern of genetic variation of five sequenced isolates of P. vivax and its divergence with two closely related species, Plasmodium cynomolgi and Plasmodium knowlesi, using a set of orthologous genes. In contrast to Plasmodium falciparum, the parasite that causes the most lethal form of human malaria, we did not find significant constraints on the evolution of synonymous sites genome wide in P. vivax. The comparative analysis of polymorphism and divergence across loci allowed us to identify 87 genes with patterns consistent with positive selection, including genes involved in the "exportome" of P. vivax, which are potentially involved in evasion of the host immune system. Nevertheless, we have found a pattern of polymorphism genome wide that is consistent with a significant amount of constraint on the replacement changes and prevalent negative selection. Our analyses also show that silent polymorphism tends to be larger toward the ends of the chromosomes, where many genes involved in antigenicity are located, suggesting that natural selection acts not only by shaping the patterns of variation within the genes but it also affects genome organization.
间日疟原虫是非洲以外最常见的人类疟原虫。然而,旨在鉴定具有与自然选择一致特征的基因的研究却很少见。在此,我们使用一组直系同源基因,对五个已测序的间日疟原虫分离株的遗传变异模式及其与两个密切相关物种——食蟹猴疟原虫和诺氏疟原虫的分化进行了比较分析。与导致人类最致命形式疟疾的恶性疟原虫不同,我们并未在间日疟原虫全基因组的同义位点进化上发现显著限制。对各基因座的多态性和分化进行比较分析,使我们能够鉴定出87个具有与正选择一致模式的基因,包括参与间日疟原虫“输出组”的基因,这些基因可能参与逃避宿主免疫系统。尽管如此,我们发现全基因组的多态性模式与对替换变化的大量限制和普遍的负选择一致。我们的分析还表明,沉默多态性在染色体末端往往更大,而许多参与抗原性的基因位于染色体末端,这表明自然选择不仅通过塑造基因内的变异模式起作用,而且还影响基因组组织。