Brue Thierry, Quentien Marie-Hélène, Khetchoumian Konstantin, Bensa Marco, Capo-Chichi José-Mario, Delemer Brigitte, Balsalobre Aurelio, Nassif Christina, Papadimitriou Dimitris T, Pagnier Anne, Hasselmann Caroline, Patry Lysanne, Schwartzentruber Jeremy, Souchon Pierre-François, Takayasu Shinobu, Enjalbert Alain, Van Vliet Guy, Majewski Jacek, Drouin Jacques, Samuels Mark E
Aix-Marseille University, Centre de Recherche en Neurobiologie et Neurophysiologie de Marseille (CRN2M), Centre National de la Recherche Scientifique, Unité Mixte de Recherche 7286, Faculté de Médecine de Marseille, 13344, Marseille, France.
Assistance Publique-Hôpitaux de Marseille (APHM), Department of Endocrinology, Centre de Référence des Maladies Rares d'Origine Hypophysaire, Hôpital de la Timone, 13005, Marseille, France.
BMC Med Genet. 2014 Dec 19;15:139. doi: 10.1186/s12881-014-0139-9.
DAVID syndrome is a rare condition combining anterior pituitary hormone deficiency with common variable immunodeficiency. NFKB2 mutations have recently been identified in patients with ACTH and variable immunodeficiency. A similar mutation was previously found in Nfkb2 in the immunodeficient Lym1 mouse strain, but the effect of the mutation on endocrine function was not evaluated.
We ascertained six unrelated DAVID syndrome families. We performed whole exome and traditional Sanger sequencing to search for causal genes. Lym1 mice were examined for endocrine developmental anomalies.
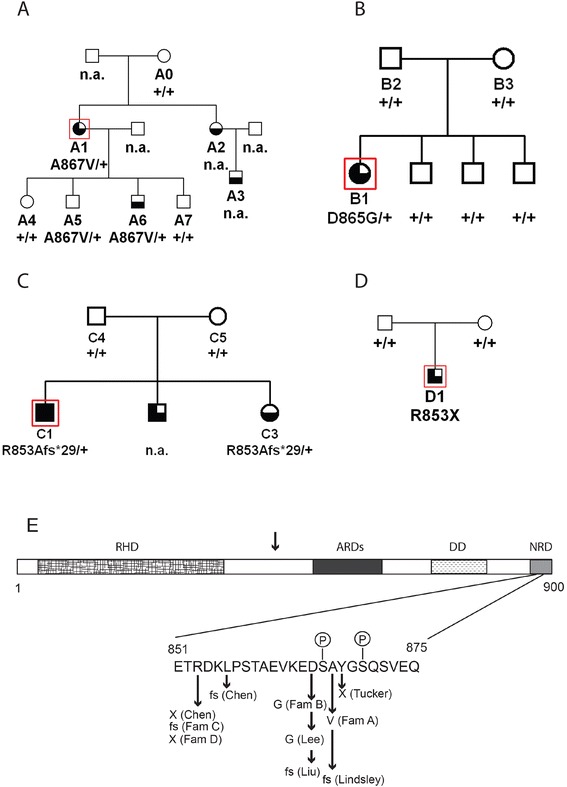
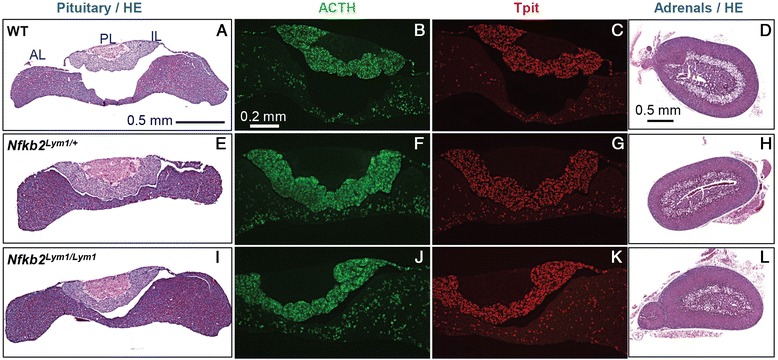
Mutations in the NFKB2 gene were identified in three of our families through whole exome sequencing, and in a fourth by direct Sanger sequencing. De novo origin of the mutations could be demonstrated in three of the families. All mutations lie near the C-terminus of the protein-coding region, near signals required for processing of NFΚB2 protein by the alternative pathway. Two of the probands had anatomical pituitary anomalies, and one had growth and thyroid hormone as well as ACTH deficiency; these findings have not been previously reported. Two children of one of the probands carried the mutation and have to date exhibited only an immune phenotype. No mutations were found near the C-terminus of NFKB2 in the remaining two probands; whole exome sequencing has been performed for one of these. Lym1 mice, carrying a similar Nfkb2 C-terminal mutation, showed normal pituitary anatomy and expression of proopiomelanocortin (POMC).
We confirm previous findings that mutations near the C-terminus of NFKB2 cause combined endocrine and immunodeficiencies. De novo status of the mutations was confirmed in all cases for which both parents were available. The mutations are consistent with a dominant gain-of-function effect, generating an unprocessed NFKB2 super-repressor protein. We expand the potential phenotype of such NFKB2 mutations to include additional pituitary hormone deficiencies as well as anatomical pituitary anomalies. The lack of an observable endocrine phenotype in Lym1 mice suggests that the endocrine component of DAVID syndrome is either not due to a direct role of NFKB pathways on pituitary development, or else that human and mouse pituitary development differ in its requirements for NFKB pathway function.
大卫综合征是一种罕见病症,合并垂体前叶激素缺乏和常见变异型免疫缺陷。最近在促肾上腺皮质激素(ACTH)缺乏和变异型免疫缺陷患者中发现了NFKB2基因突变。此前在免疫缺陷的Lym1小鼠品系的Nfkb2中发现了类似突变,但未评估该突变对内分泌功能的影响。
我们确定了6个无血缘关系的大卫综合征家族。我们进行了全外显子组测序和传统桑格测序以寻找致病基因。对Lym1小鼠进行内分泌发育异常检查。
通过全外显子组测序在我们的3个家族中鉴定出NFKB2基因突变,在第4个家族中通过直接桑格测序鉴定出该突变。在其中3个家族中证实了突变的新生性。所有突变均位于蛋白质编码区的C末端附近,靠近通过替代途径加工NFΚB2蛋白所需的信号。2名先证者存在垂体解剖学异常,1名先证者存在生长激素、甲状腺激素以及促肾上腺皮质激素缺乏;这些发现此前未见报道。1名先证者的2名子女携带该突变,迄今为止仅表现出免疫表型。在其余2名先证者的NFKB2基因C末端附近未发现突变;已对其中1名进行了全外显子组测序。携带类似Nfkb2 C末端突变的Lym1小鼠垂体解剖结构和阿黑皮素原(POMC)表达正常。
我们证实了先前的发现,即NFKB2基因C末端附近的突变会导致内分泌和免疫缺陷合并出现。在所有父母均可用的病例中均证实了突变的新生性。这些突变与显性功能获得效应一致,产生未加工的NFKB2超级抑制蛋白。我们将此类NFKB2突变的潜在表型扩展至包括其他垂体激素缺乏以及垂体解剖学异常。Lym1小鼠缺乏可观察到的内分泌表型,这表明大卫综合征的内分泌成分要么不是由于NFKB信号通路对垂体发育的直接作用,要么是人与小鼠垂体发育对NFKB信号通路功能的需求不同。