Goodswen Stephen J, Kennedy Paul J, Ellis John T
School of Medical and Molecular Sciences, University of Technology Sydney (UTS), Ultimo, NSW, Australia.
School of Software, Faculty of Engineering and Information Technology and the Centre for Quantum Computation and Intelligent Systems at the University of Technology Sydney (UTS), Ultimo, NSW, Australia.
PLoS One. 2014 Dec 29;9(12):e115745. doi: 10.1371/journal.pone.0115745. eCollection 2014.
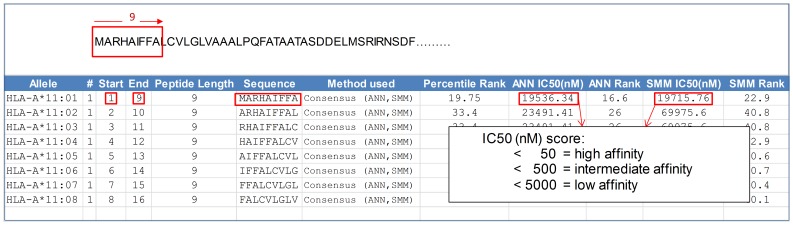
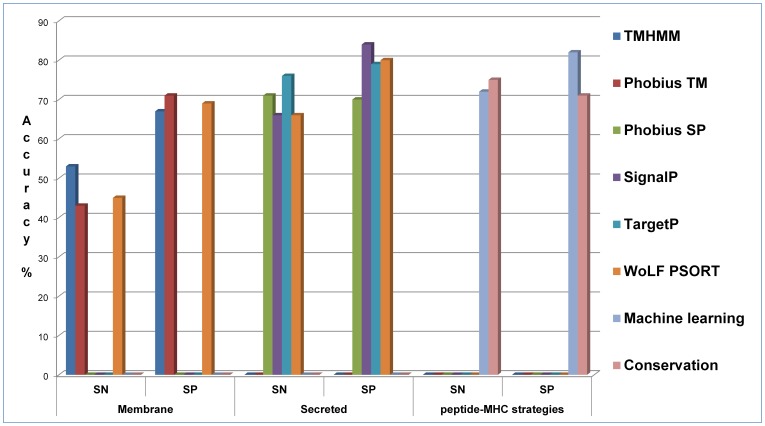
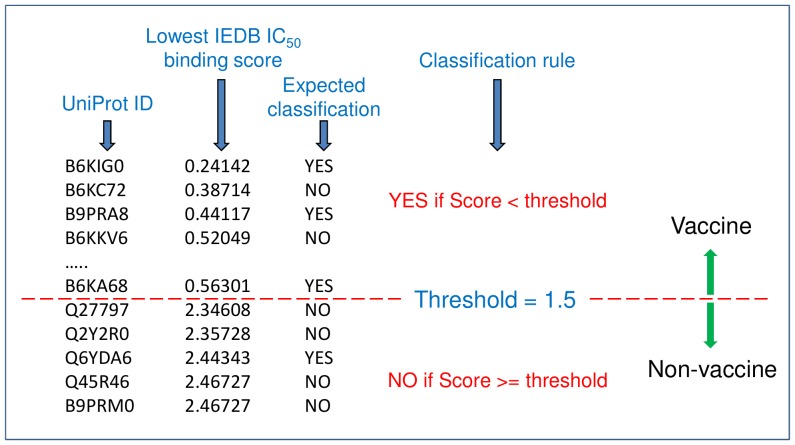
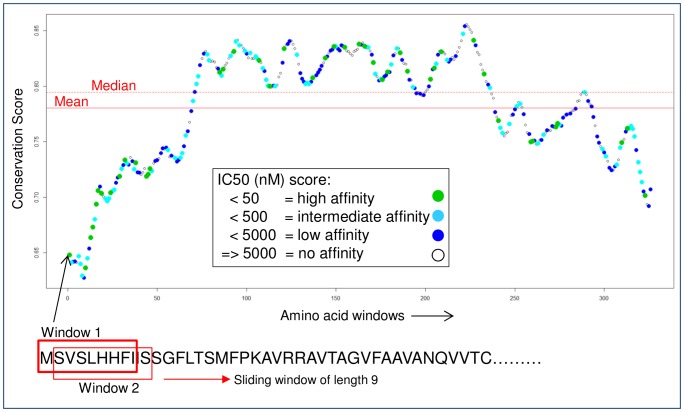
Given thousands of proteins constituting a eukaryotic pathogen, the principal objective for a high-throughput in silico vaccine discovery pipeline is to select those proteins worthy of laboratory validation. Accurate prediction of T-cell epitopes on protein antigens is one crucial piece of evidence that would aid in this selection. Prediction of peptides recognised by T-cell receptors have to date proved to be of insufficient accuracy. The in silico approach is consequently reliant on an indirect method, which involves the prediction of peptides binding to major histocompatibility complex (MHC) molecules. There is no guarantee nevertheless that predicted peptide-MHC complexes will be presented by antigen-presenting cells and/or recognised by cognate T-cell receptors. The aim of this study was to determine if predicted peptide-MHC binding scores could provide contributing evidence to establish a protein's potential as a vaccine. Using T-Cell MHC class I binding prediction tools provided by the Immune Epitope Database and Analysis Resource, peptide binding affinity to 76 common MHC I alleles were predicted for 160 Toxoplasma gondii proteins: 75 taken from published studies represented proteins known or expected to induce T-cell immune responses and 85 considered less likely vaccine candidates. The results show there is no universal set of rules that can be applied directly to binding scores to distinguish a vaccine from a non-vaccine candidate. We present, however, two proposed strategies exploiting binding scores that provide supporting evidence that a protein is likely to induce a T-cell immune response-one using random forest (a machine learning algorithm) with a 72% sensitivity and 82.4% specificity and the other, using amino acid conservation scores with a 74.6% sensitivity and 70.5% specificity when applied to the 160 benchmark proteins. More importantly, the binding score strategies are valuable evidence contributors to the overall in silico vaccine discovery pool of evidence.
真核病原体由数千种蛋白质构成,高通量计算机辅助疫苗发现流程的主要目标是筛选出那些值得进行实验室验证的蛋白质。准确预测蛋白质抗原上的T细胞表位是有助于这一筛选的关键证据之一。迄今为止,T细胞受体识别的肽段预测准确性不足。因此,计算机辅助方法依赖于一种间接方法,即预测与主要组织相容性复合体(MHC)分子结合的肽段。然而,无法保证预测的肽-MHC复合物会由抗原呈递细胞呈递和/或被同源T细胞受体识别。本研究的目的是确定预测的肽-MHC结合分数是否能为确定一种蛋白质作为疫苗的潜力提供有力证据。利用免疫表位数据库和分析资源提供的T细胞MHC I类结合预测工具,对160种刚地弓形虫蛋白与76种常见MHC I等位基因的肽结合亲和力进行了预测:75种取自已发表研究,代表已知或预期能诱导T细胞免疫反应的蛋白质,85种被认为是不太可能成为疫苗候选物的蛋白质。结果表明,没有一套通用规则可直接应用于结合分数以区分疫苗候选物和非疫苗候选物。然而,我们提出了两种利用结合分数的策略,它们为一种蛋白质可能诱导T细胞免疫反应提供了支持证据——一种使用随机森林(一种机器学习算法),灵敏度为72%,特异性为82.4%;另一种使用氨基酸保守分数,应用于160种基准蛋白质时,灵敏度为74.6%,特异性为70.5%。更重要的是,结合分数策略是计算机辅助疫苗发现证据库的重要证据来源。