Allen-Brady Kristina, Cannon-Albright Lisa A, Farnham James M, Norton Peggy A
Division of Genetic Epidemiology, Department of Internal Medicine, University of Utah School of Medicine, Salt Lake City, UT.
Division of Genetic Epidemiology, Department of Internal Medicine, University of Utah School of Medicine, Salt Lake City, UT; Research and Development Service, George E. Wahlen Department of Veterans Affairs Medical Center, Salt Lake City, UT.
Am J Obstet Gynecol. 2015 Jun;212(6):771.e1-7. doi: 10.1016/j.ajog.2014.12.037. Epub 2014 Dec 31.
We conducted a genomewide linkage analysis to identify pelvic organ prolapse (POP) predisposition genes using a resource of high-risk POP pedigrees.
Cases are defined as women who reported bothersome symptoms of POP based on standardized symptom questions (Pelvic Floor Distress Inventory, moderately or quite bothered), and/or received treatment for POP documented in medical records. Our complete pedigree resource contains 299 familial POP cases in 83 high-risk pedigrees. Genotype data were obtained from Illumina HumanHap550, 610Q, the Human1M-Duo, Human Omni1-Quad, or the Human Omni 2.5 platforms. A set of single nucleotide polymorphism markers common to all platforms was identified and markers in high linkage disequilibrium were removed. We performed a genomewide linkage analysis under general dominant and recessive models using a Markov chain, Monte Carlo linkage analysis method implemented in MCLINK (University of Utah) software. Because 70 individuals in 32 pedigrees were used in a previously published linkage analysis for a phenotype of POP requiring treatment/surgery, we also performed linkage only including the 225 newly recruited and genotyped cases in 61 pedigrees.
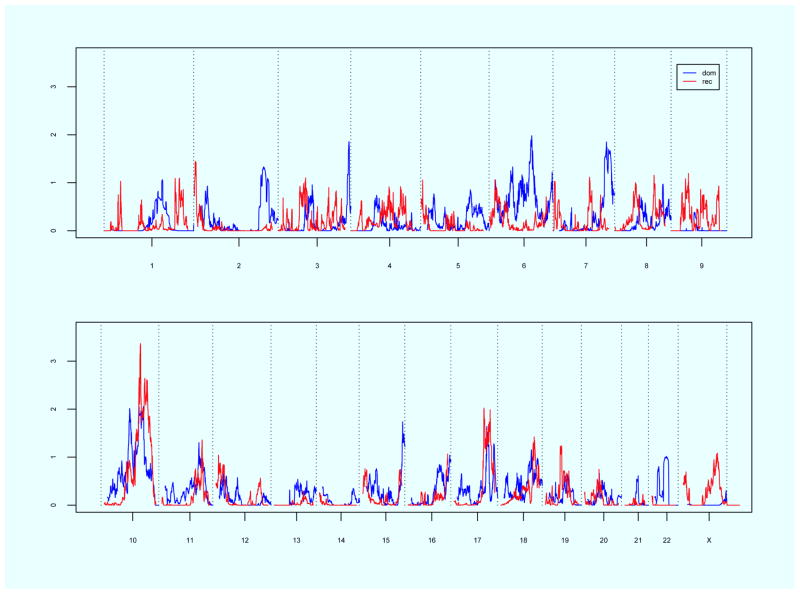
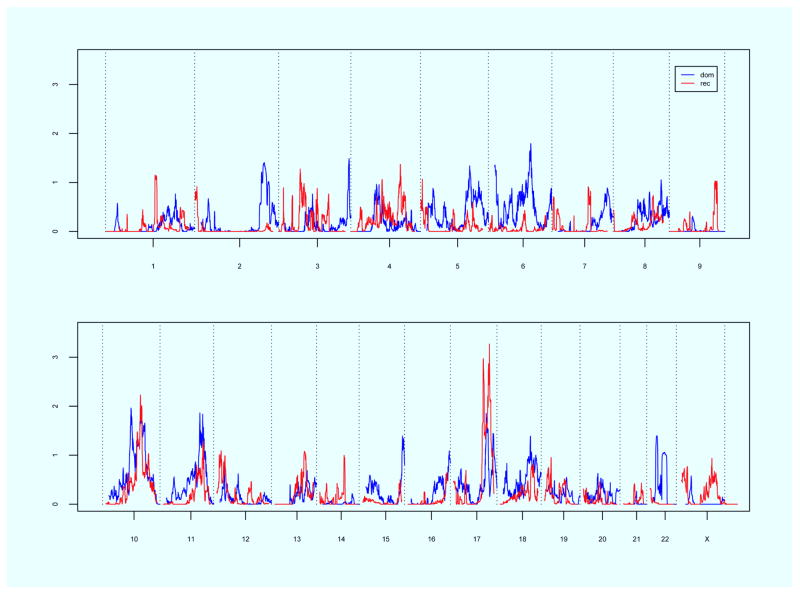
Linkage analysis using our complete pedigree resource for the loosened criteria of bothersome POP showed evidence for significant genomewide linkage on chromosome 10q24-26 (recessive model, maximum heterogeneity logarithm of odds 3.4); suggestive evidence was identified on chromosomes 6 and 17, and an additional region on chromosome 10. In the subset of only the newly recruited familial POP cases, significant evidence for genomewide linkage was observed on chromosome 17q25 (recessive model, maximum heterogeneity logarithm of odds 3.3), and suggestive evidence for linkage was observed on chromosomes 10 and 11. Neither analysis duplicated the previously published linkage evidence for the POP requiring treatment/surgery phenotype observed on chromosome 9.
While the etiology of this common condition is unknown, this study provides evidence that loci on chromosomes 10q and 17q may contribute to POP etiology.
我们利用高危盆腔器官脱垂(POP)家系资源进行全基因组连锁分析,以确定POP的易感基因。
病例定义为根据标准化症状问题(盆底困扰量表,中度或非常困扰)报告有烦人的POP症状,和/或病历中有POP治疗记录的女性。我们完整的家系资源包含83个高危家系中的299例家族性POP病例。基因型数据来自Illumina HumanHap550、610Q、Human1M-Duo、Human Omni1-Quad或Human Omni 2.5平台。确定了所有平台共有的一组单核苷酸多态性标记,并去除了处于高连锁不平衡状态的标记。我们使用MCLINK(犹他大学)软件中实现的马尔可夫链蒙特卡罗连锁分析方法,在一般显性和隐性模型下进行全基因组连锁分析。由于32个家系中的70名个体曾在先前发表的一项针对需要治疗/手术的POP表型的连锁分析中使用过,我们还进行了仅包括61个家系中225例新招募并进行基因分型的病例的连锁分析。
使用我们完整的家系资源对烦人的POP宽松标准进行连锁分析,结果显示在染色体10q24 - 26上有全基因组显著连锁的证据(隐性模型,最大异质性对数优势比为3.4);在染色体6和17以及染色体10上的另一个区域发现了提示性证据。在仅新招募的家族性POP病例子集中,在染色体17q25上观察到全基因组连锁的确凿证据(隐性模型,最大异质性对数优势比为3.3),在染色体10和11上观察到连锁的提示性证据。两项分析均未重复先前发表的在染色体9上观察到的需要治疗/手术的POP表型的连锁证据。
虽然这种常见疾病的病因尚不清楚,但本研究提供了证据表明染色体10q和17q上的基因座可能与POP病因有关。