Pearl Phillip L, Parviz Mahsa, Vogel Kara, Schreiber John, Theodore William H, Gibson K Michael
Department of Neurology, Boston Children's Hospital, Harvard Medical School, Boston, MA, USA.
Department of Experimental and Systems Pharmacology, College of Pharmacy, Washington State University, Spokane, WA, USA.
Dev Med Child Neurol. 2015 Jul;57(7):611-617. doi: 10.1111/dmcn.12668. Epub 2014 Dec 29.
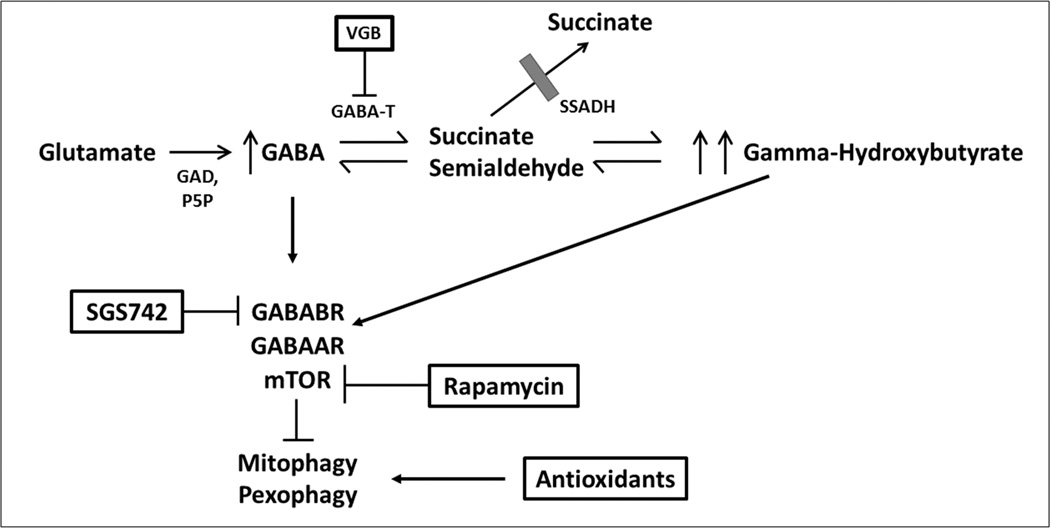
Inherited disorders of gamma-aminobutyric acid (GABA) metabolism include succinic semialdehyde dehydrogenase (SSADH) and gamma-aminobutyric acid transaminase (GABA-T) deficiencies. The clinical features, pathophysiology, diagnosis, and management of both, and an updated list of mutations in the ALDH5A1 gene, which cause SSADH deficiency, are discussed. A database of 112 individuals (71 children and adolescents, and 41 adults) indicates that developmental delay and hypotonia are the most common symptoms arising from SSADH deficiency. Furthermore, epilepsy is present in two-thirds of SSADH-deficient individuals by adulthood. Research with murine genetic models and human participants, using [ C] flumazenil positron emission tomography (FMZ-PET) and transcranial magnetic stimulation, have led to therapeutic trials, and the identification of additional disruptions to GABA metabolism. Suggestions for new therapies have arisen from findings of GABAergic effects on autophagy, with enhanced activation of the mammalian target of rapamycin (mTOR) pathway. Details of known pathogenic mutations in the ALDH5A1 gene, three of which have not previously been reported, are summarized here. Investigations into disorders of GABA metabolism provide fundamental insights into the mechanisms underlying epilepsy, and support the importance of developing biomarkers and clinical trials. Comprehensive definition of phenotypes arising as a result of deficiencies in both SSADH and GABA-T may increase our understanding of the neurophysiological consequences of a hyper-GABAergic state.
γ-氨基丁酸(GABA)代谢的遗传性疾病包括琥珀酸半醛脱氢酶(SSADH)缺乏症和γ-氨基丁酸转氨酶(GABA-T)缺乏症。本文讨论了这两种疾病的临床特征、病理生理学、诊断和管理,以及导致SSADH缺乏症的ALDH5A1基因突变的最新列表。一个包含112名个体(71名儿童和青少年以及41名成年人)的数据库表明,发育迟缓与肌张力减退是SSADH缺乏症最常见的症状。此外,到成年期时,三分之二的SSADH缺乏症患者患有癫痫。利用[C]氟马西尼正电子发射断层扫描(FMZ-PET)和经颅磁刺激对小鼠遗传模型和人类受试者进行的研究已促成了治疗试验,并发现了GABA代谢的其他紊乱情况。对自噬的GABA能作用的研究结果提出了新的治疗建议,同时哺乳动物雷帕霉素靶蛋白(mTOR)途径的激活增强。本文总结了ALDH5A1基因中已知致病突变的详细信息,其中三个突变此前未曾报道。对GABA代谢紊乱的研究为癫痫的潜在机制提供了基本见解,并支持开发生物标志物和开展临床试验的重要性。全面定义由SSADH和GABA-T缺乏导致的表型可能会增进我们对高GABA能状态的神经生理后果的理解。