Stefan Diana, Pinel Corinne, Pinhal Stéphane, Cinquemani Eugenio, Geiselmann Johannes, de Jong Hidde
INRIA Grenoble - Rhône-Alpes, Grenoble, France; Laboratoire Interdisciplinaire de Physique (LIPhy, CNRS UMR 5588), Université Joseph Fourier, Grenoble, France.
INRIA Grenoble - Rhône-Alpes, Grenoble, France.
PLoS Comput Biol. 2015 Jan 15;11(1):e1004028. doi: 10.1371/journal.pcbi.1004028. eCollection 2015 Jan.
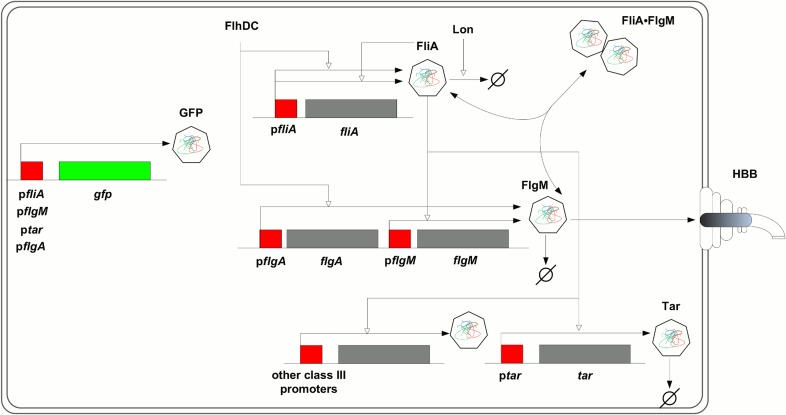
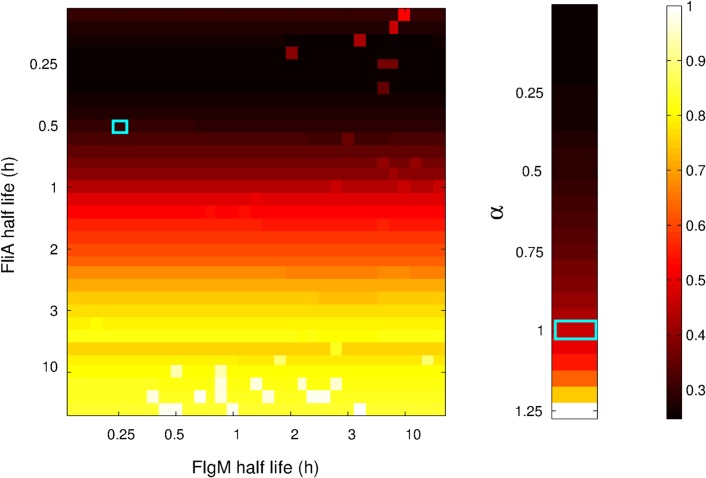
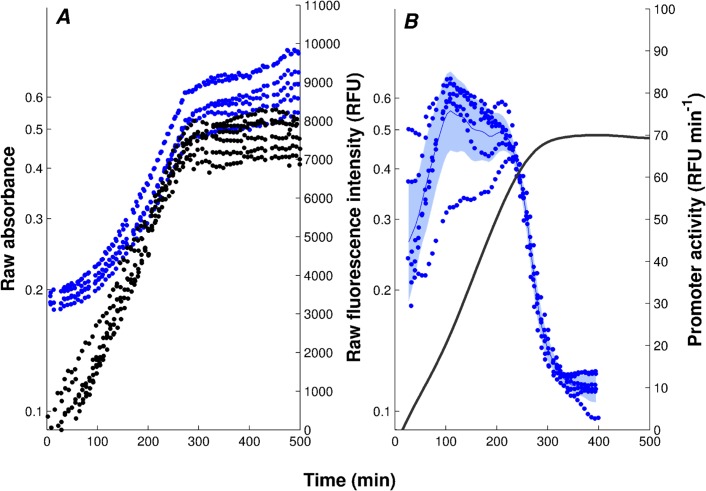
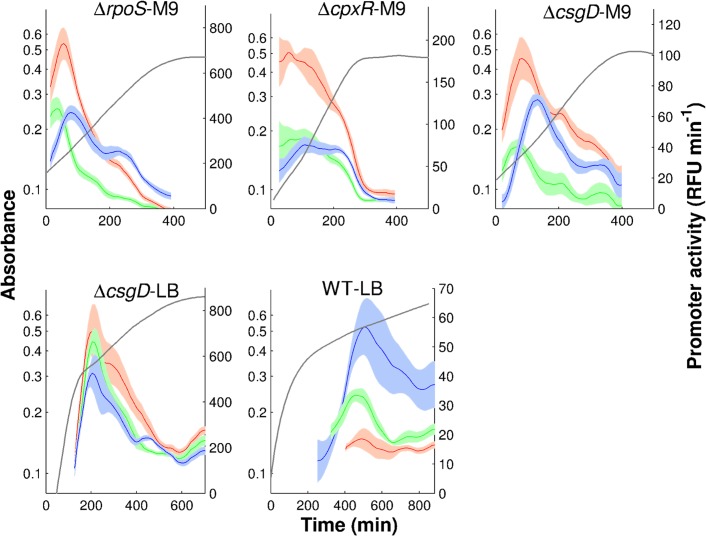
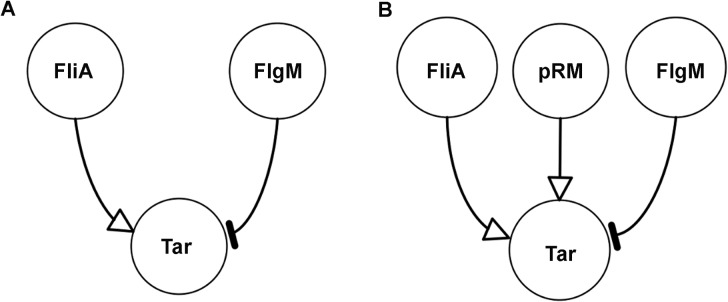
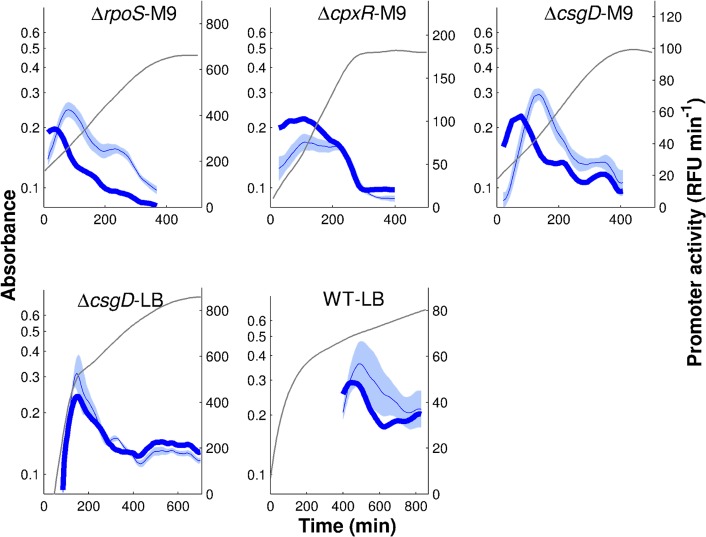
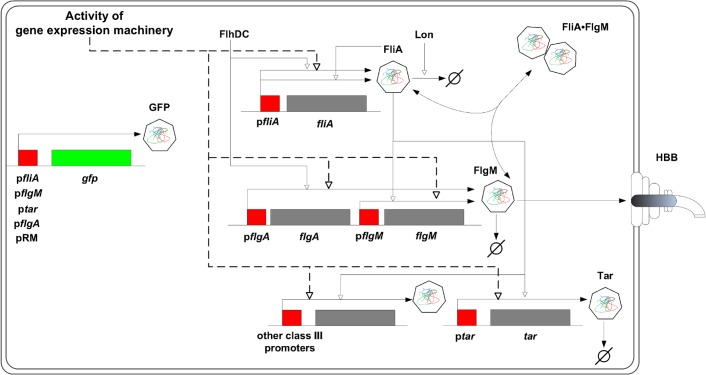
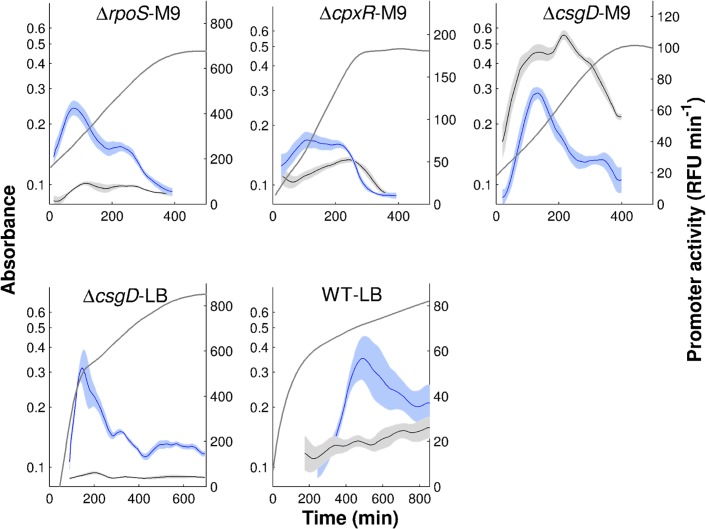
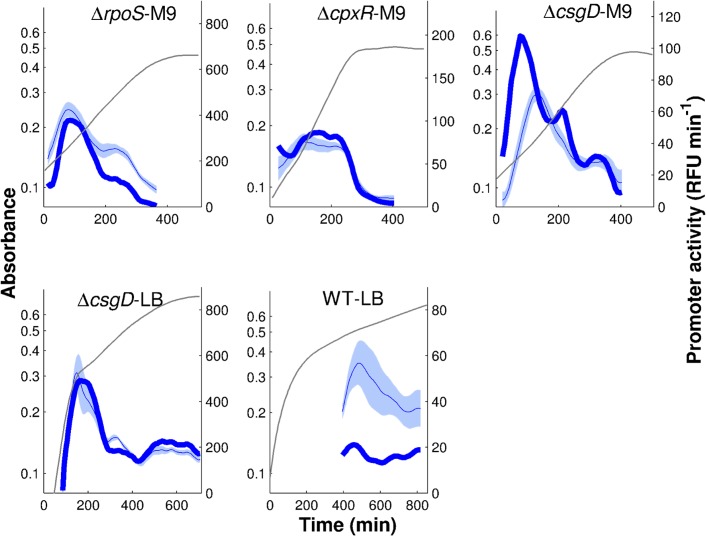
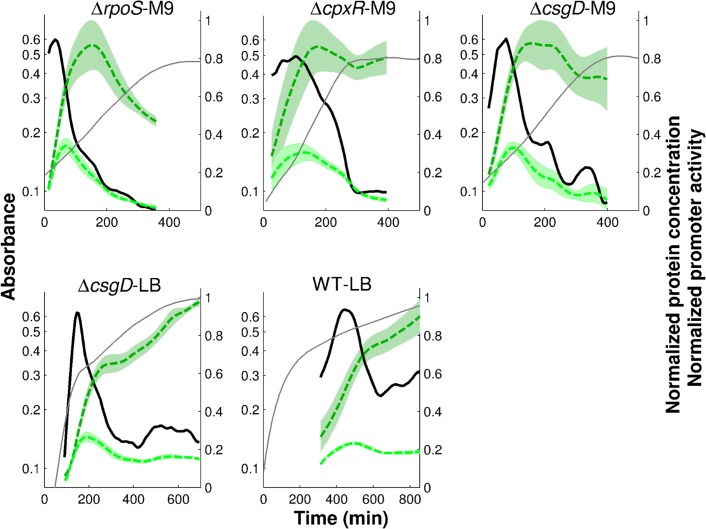
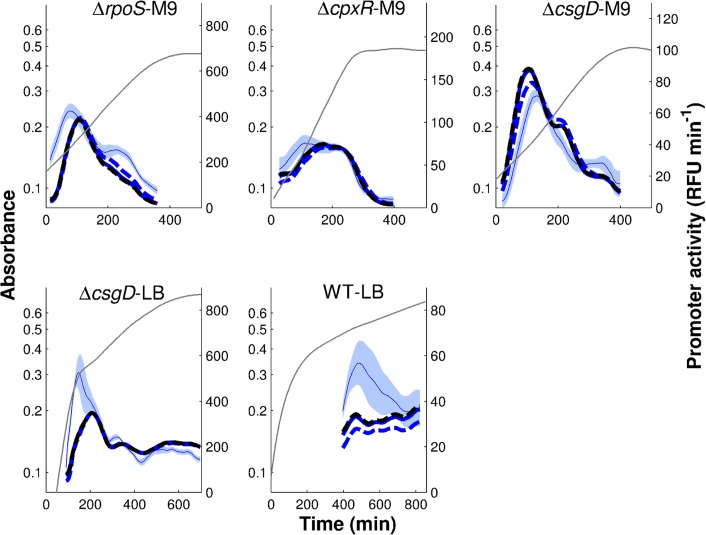
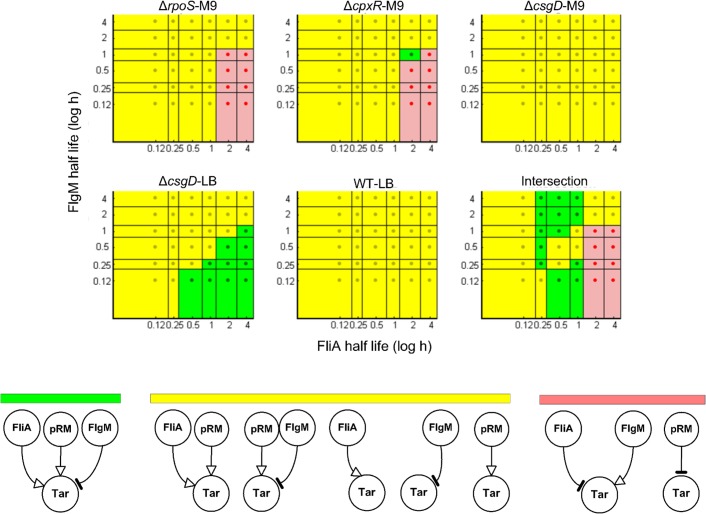
The inference of regulatory interactions and quantitative models of gene regulation from time-series transcriptomics data has been extensively studied and applied to a range of problems in drug discovery, cancer research, and biotechnology. The application of existing methods is commonly based on implicit assumptions on the biological processes under study. First, the measurements of mRNA abundance obtained in transcriptomics experiments are taken to be representative of protein concentrations. Second, the observed changes in gene expression are assumed to be solely due to transcription factors and other specific regulators, while changes in the activity of the gene expression machinery and other global physiological effects are neglected. While convenient in practice, these assumptions are often not valid and bias the reverse engineering process. Here we systematically investigate, using a combination of models and experiments, the importance of this bias and possible corrections. We measure in real time and in vivo the activity of genes involved in the FliA-FlgM module of the E. coli motility network. From these data, we estimate protein concentrations and global physiological effects by means of kinetic models of gene expression. Our results indicate that correcting for the bias of commonly-made assumptions improves the quality of the models inferred from the data. Moreover, we show by simulation that these improvements are expected to be even stronger for systems in which protein concentrations have longer half-lives and the activity of the gene expression machinery varies more strongly across conditions than in the FliA-FlgM module. The approach proposed in this study is broadly applicable when using time-series transcriptome data to learn about the structure and dynamics of regulatory networks. In the case of the FliA-FlgM module, our results demonstrate the importance of global physiological effects and the active regulation of FliA and FlgM half-lives for the dynamics of FliA-dependent promoters.
从时间序列转录组学数据推断调控相互作用和基因调控的定量模型已得到广泛研究,并应用于药物发现、癌症研究和生物技术等一系列问题。现有方法的应用通常基于对所研究生物过程的隐含假设。首先,转录组学实验中获得的mRNA丰度测量值被视为蛋白质浓度的代表。其次,假设观察到的基因表达变化仅归因于转录因子和其他特定调节因子,而忽略了基因表达机制活性的变化和其他全局生理效应。虽然这些假设在实践中很方便,但往往无效,并使逆向工程过程产生偏差。在这里,我们结合模型和实验系统地研究了这种偏差的重要性以及可能的校正方法。我们实时且在体内测量了大肠杆菌运动网络中FliA-FlgM模块相关基因的活性。从这些数据中,我们通过基因表达动力学模型估计蛋白质浓度和全局生理效应。我们的结果表明,校正常见假设的偏差可提高从数据推断出的模型质量。此外,我们通过模拟表明,对于蛋白质浓度半衰期更长且基因表达机制活性在不同条件下变化比FliA-FlgM模块更强烈的系统,这些改进预计会更强。本研究中提出的方法在使用时间序列转录组数据了解调控网络的结构和动态时具有广泛的适用性。对于FliA-FlgM模块,我们的结果证明了全局生理效应以及FliA和FlgM半衰期的主动调节对FliA依赖性启动子动态的重要性。