Dey Siddharth S, Kester Lennart, Spanjaard Bastiaan, Bienko Magda, van Oudenaarden Alexander
Hubrecht Institute-KNAW (Royal Netherlands Academy of Arts and Sciences), Utrecht, The Netherlands.
University Medical Center Utrecht, Cancer Genomics Netherlands, Utrecht, The Netherlands.
Nat Biotechnol. 2015 Mar;33(3):285-289. doi: 10.1038/nbt.3129. Epub 2015 Jan 19.
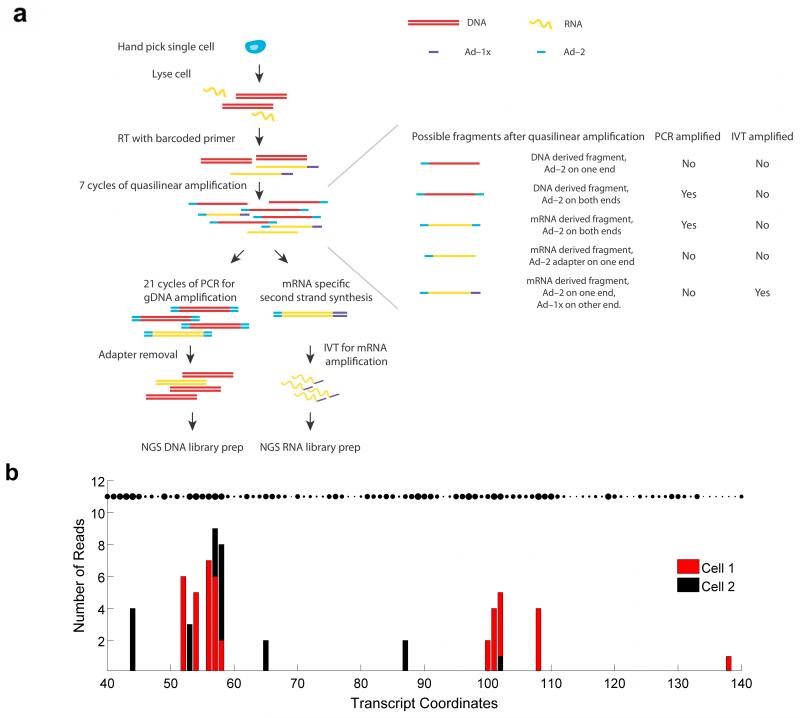
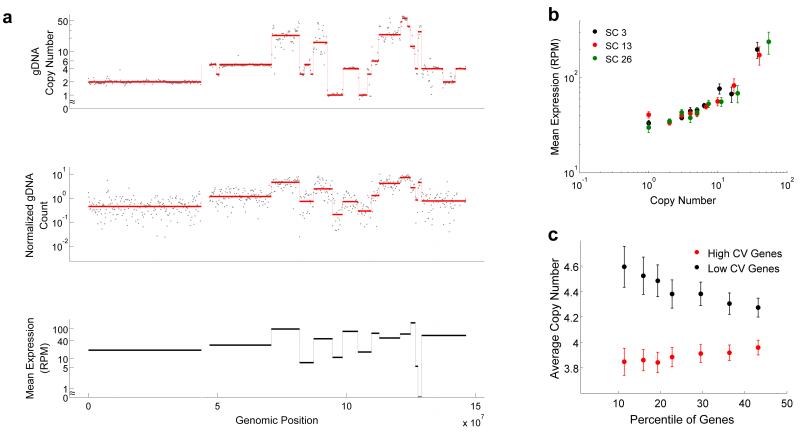
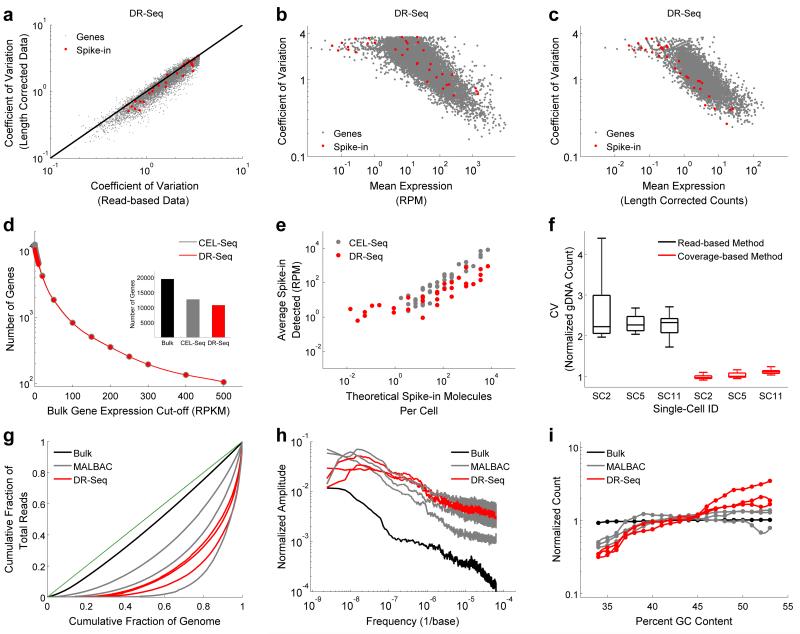
Single-cell genomics and single-cell transcriptomics have emerged as powerful tools to study the biology of single cells at a genome-wide scale. However, a major challenge is to sequence both genomic DNA and mRNA from the same cell, which would allow direct comparison of genomic variation and transcriptome heterogeneity. We describe a quasilinear amplification strategy to quantify genomic DNA and mRNA from the same cell without physically separating the nucleic acids before amplification. We show that the efficiency of our integrated approach is similar to existing methods for single-cell sequencing of either genomic DNA or mRNA. Further, we find that genes with high cell-to-cell variability in transcript numbers generally have lower genomic copy numbers, and vice versa, suggesting that copy number variations may drive variability in gene expression among individual cells. Applications of our integrated sequencing approach could range from gaining insights into cancer evolution and heterogeneity to understanding the transcriptional consequences of copy number variations in healthy and diseased tissues.
单细胞基因组学和单细胞转录组学已成为在全基因组范围内研究单细胞生物学的强大工具。然而,一个主要挑战是对同一细胞的基因组DNA和mRNA进行测序,这将允许直接比较基因组变异和转录组异质性。我们描述了一种准线性扩增策略,用于在扩增前不物理分离核酸的情况下定量同一细胞中的基因组DNA和mRNA。我们表明,我们的综合方法的效率与现有的基因组DNA或mRNA单细胞测序方法相似。此外,我们发现转录本数量在细胞间变化较大的基因通常基因组拷贝数较低,反之亦然,这表明拷贝数变异可能驱动个体细胞间基因表达的变化。我们的综合测序方法的应用范围可以从深入了解癌症进化和异质性到理解健康和患病组织中拷贝数变异的转录后果。