Mertes Florian, Lichtner Björn, Kuhl Heiner, Blattner Mirjam, Otte Jörg, Wruck Wasco, Timmermann Bernd, Lehrach Hans, Adjaye James
Department of Vertebrate Genomics, Max Planck Institute for Molecular Genetics, Ihnestr. 63-73, 14195, Berlin, Germany.
Molecular Exposomics, Helmholtz Zentrum München, Ingolstädter Landstr. 1, 85764, Neuherberg, Germany.
BMC Genomics. 2015 Nov 12;16:925. doi: 10.1186/s12864-015-2025-z.
Next Generation Sequencing has proven to be an exceptionally powerful tool in the field of genomics and transcriptomics. With recent development it is nowadays possible to analyze ultra-low input sample material down to single cells. Nevertheless, investigating such sample material often limits the analysis to either the genome or transcriptome. We describe here a combined analysis of both types of nucleic acids from the same sample material.
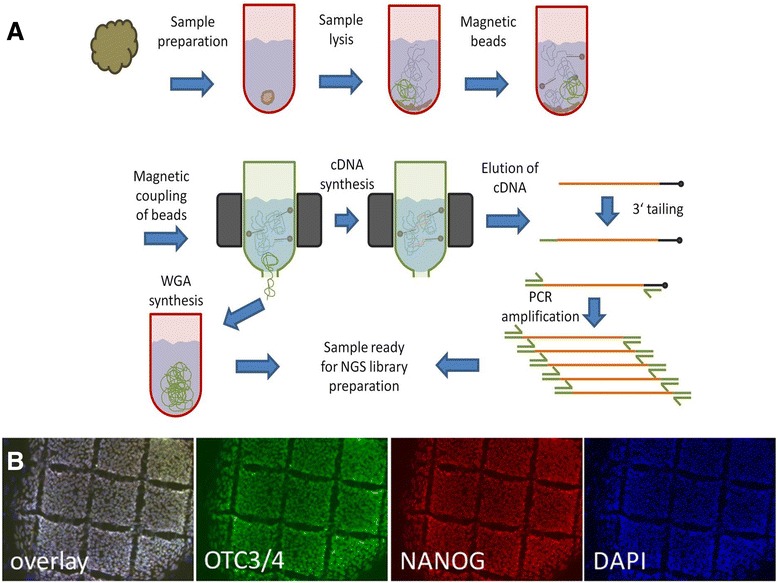
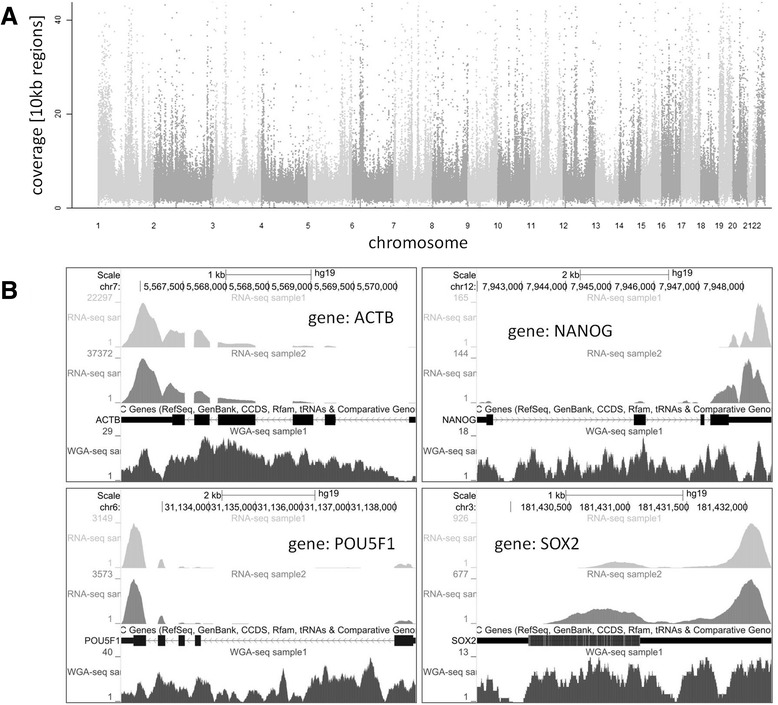
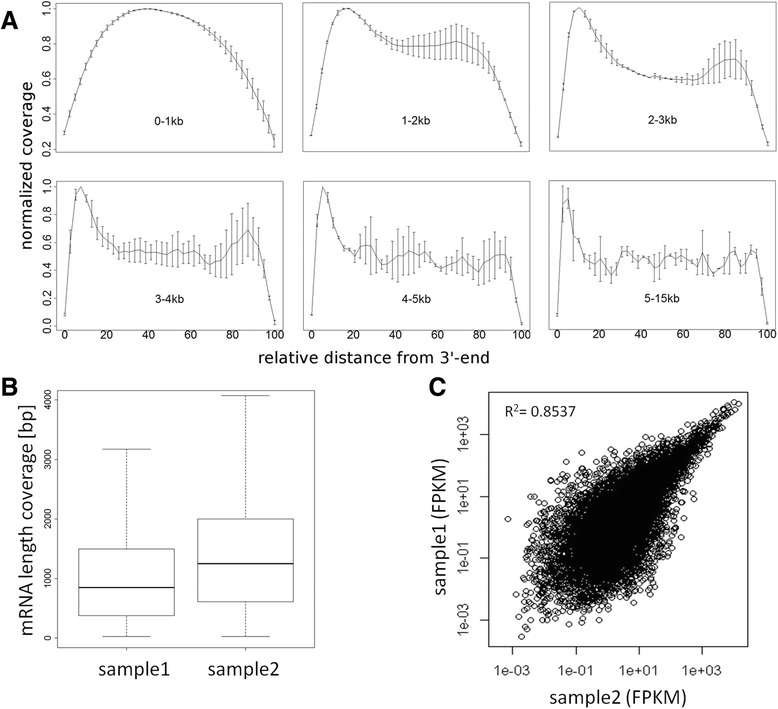
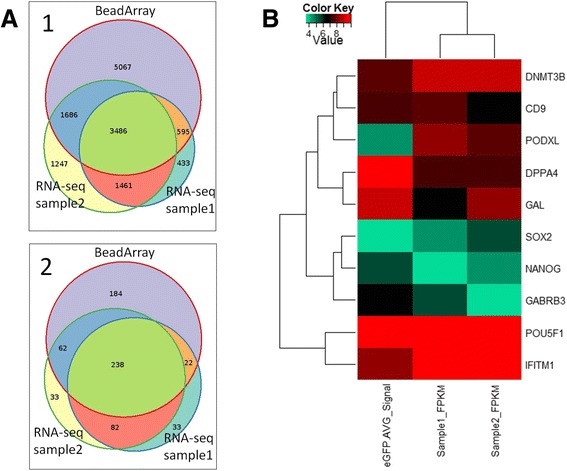
The method described enables the combined preparation of amplified cDNA as well as amplified whole-genome DNA from an ultra-low input sample material derived from a sub-colony of in-vitro cultivated human embryonic stem cells. cDNA is prepared by the application of oligo-dT coupled magnetic beads for mRNA capture, first strand synthesis and 3'-tailing followed by PCR. Whole-genome amplified DNA is prepared by Phi29 mediated amplification. Illumina sequencing is applied to short fragment libraries prepared from the amplified samples.
We developed a protocol which enables the combined analysis of the genome as well as the transcriptome by Next Generation Sequencing from ultra-low input samples. The protocol was evaluated by sequencing sub-colony structures from human embryonic stem cells containing 150 to 200 cells. The method can be adapted to any available sequencing system.
To our knowledge, this is the first report where sub-colonies of human embryonic stem cells have been analyzed both at the genomic as well as transcriptome level. The method of this proof of concept study may find useful practical applications for cases where only a limited number of cells are available, e.g. for tissues samples from biopsies, tumor spheres, circulating tumor cells and cells from early embryonic development. The results we present demonstrate that a combined analysis of genomic DNA and messenger RNA from ultra-low input samples is feasible and can readily be applied to other cellular systems with limited material available.
下一代测序已被证明是基因组学和转录组学领域中一种极其强大的工具。随着近期的发展,如今能够分析低至单细胞的超低输入样本材料。然而,研究此类样本材料通常会将分析局限于基因组或转录组。我们在此描述对来自同一样本材料的两种核酸类型进行联合分析。
所描述的方法能够从体外培养的人类胚胎干细胞亚克隆的超低输入样本材料中联合制备扩增的cDNA以及扩增的全基因组DNA。通过应用与寡聚dT偶联的磁珠捕获mRNA、进行第一链合成和3'加尾,随后进行PCR来制备cDNA。通过Phi29介导的扩增制备全基因组扩增DNA。将Illumina测序应用于从扩增样本制备的短片段文库。
我们开发了一种方案,该方案能够通过下一代测序对超低输入样本的基因组和转录组进行联合分析。通过对包含150至200个细胞的人类胚胎干细胞亚克隆结构进行测序来评估该方案。该方法可适用于任何可用的测序系统。
据我们所知,这是第一份关于在基因组和转录组水平对人类胚胎干细胞亚克隆进行分析的报告。这一概念验证研究的方法可能在仅有有限数量细胞可用的情况下找到有用的实际应用,例如对于活检组织样本、肿瘤球、循环肿瘤细胞以及早期胚胎发育的细胞。我们展示的结果表明,对超低输入样本的基因组DNA和信使RNA进行联合分析是可行的,并且可以很容易地应用于其他材料有限的细胞系统。