Navarro Jean-Marc, Touzart Aurore, Pradel Lydie C, Loosveld Marie, Koubi Myriam, Fenouil Romain, Le Noir Sandrine, Maqbool Muhammad Ahmad, Morgado Ester, Gregoire Claude, Jaeger Sebastien, Mamessier Emilie, Pignon Charles, Hacein-Bey-Abina Salima, Malissen Bernard, Gut Marta, Gut Ivo G, Dombret Hervé, Macintyre Elizabeth A, Howe Steven J, Gaspar H Bobby, Thrasher Adrian J, Ifrah Norbert, Payet-Bornet Dominique, Duprez Estelle, Andrau Jean-Christophe, Asnafi Vahid, Nadel Bertrand
1] Center of Immunology of Marseille Luminy, Aix-Marseille University, Parc Scientifique de Luminy case 906, 13288 Marseille, France [2] INSERM U1104, 13288 Marseille, France [3] CNRS UMR7280, 13288 Marseille, France.
Université Paris Descartes Sorbonne Cité, Institut Necker-Enfants Malades (INEM), Institut national de recherche médicale (INSERM) U1151, and Laboratory of Onco-Hematology, Assistance Publique-Hôpitaux de Paris (AP-HP), Hôpital Necker Enfants-Malades, 75015 Paris, France.
Nat Commun. 2015 Jan 23;6:6094. doi: 10.1038/ncomms7094.
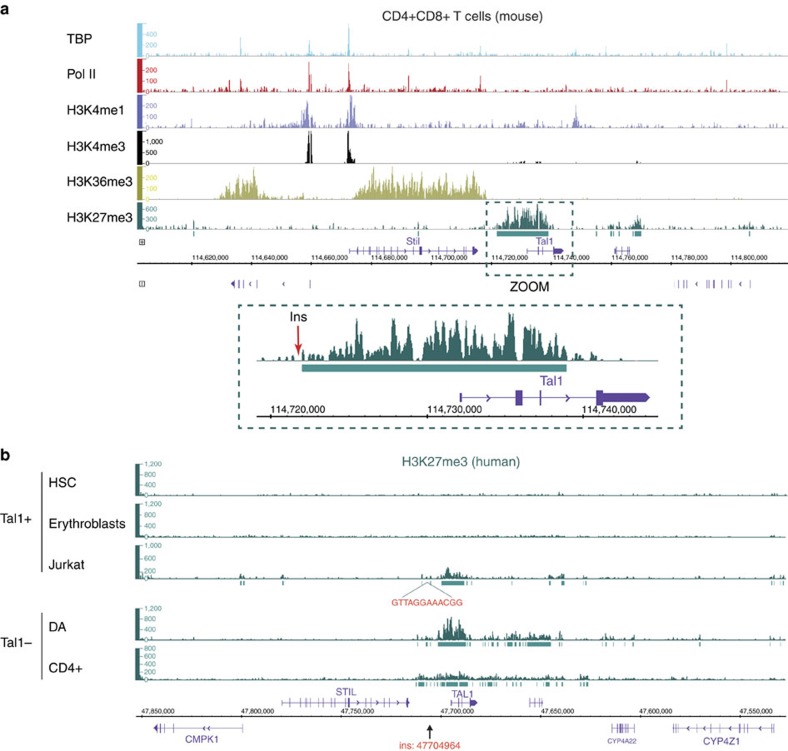
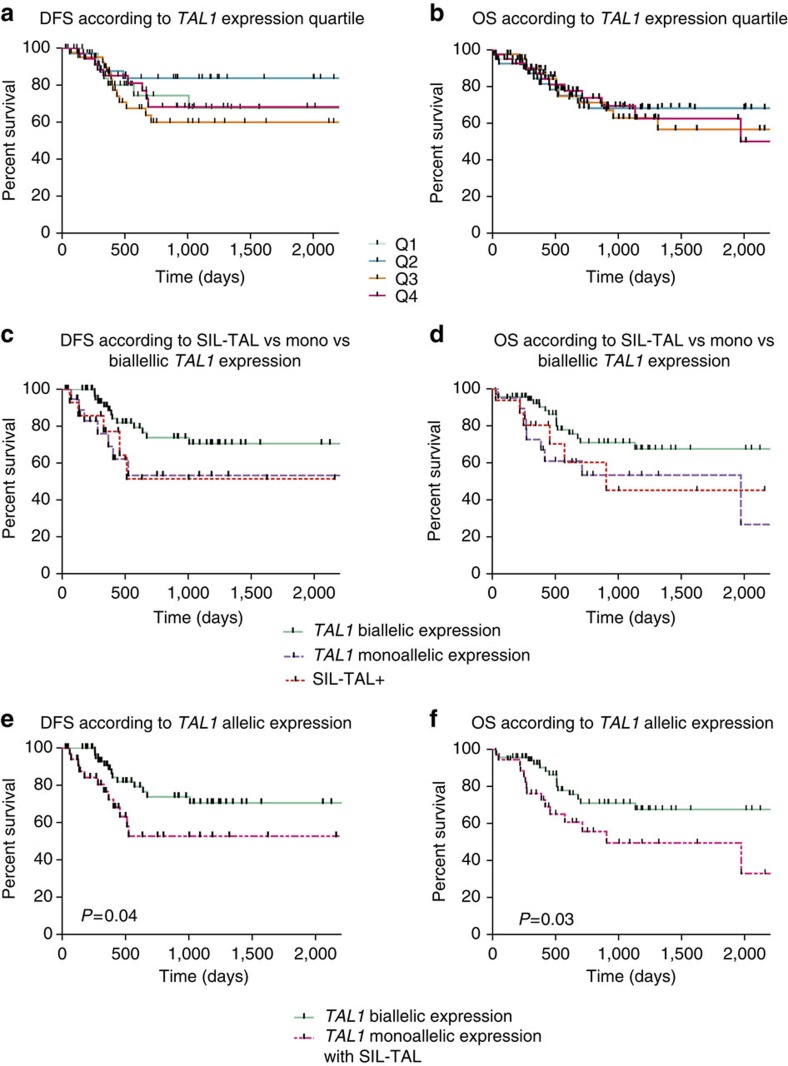
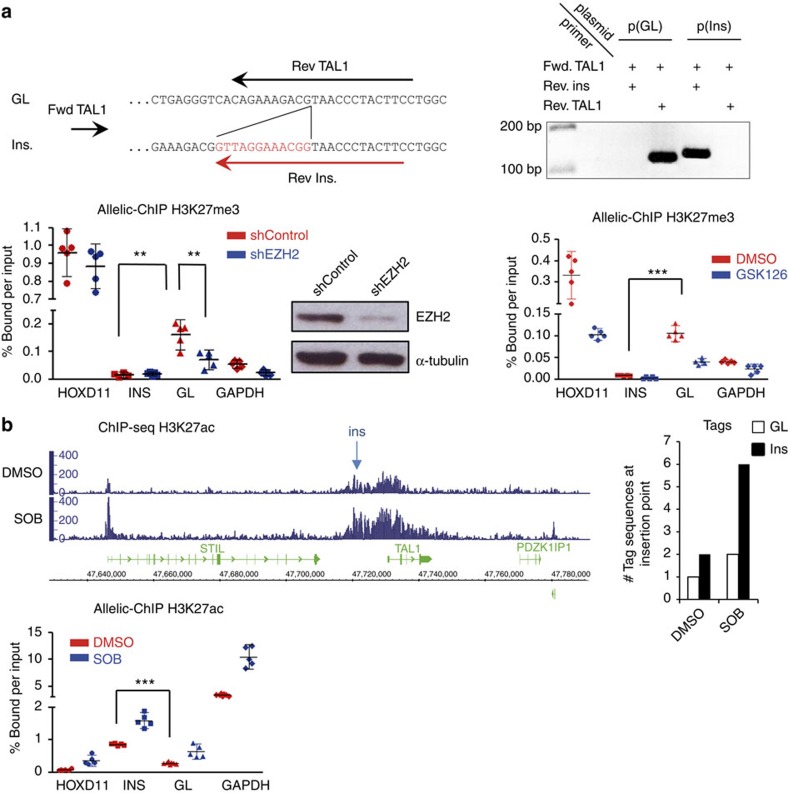
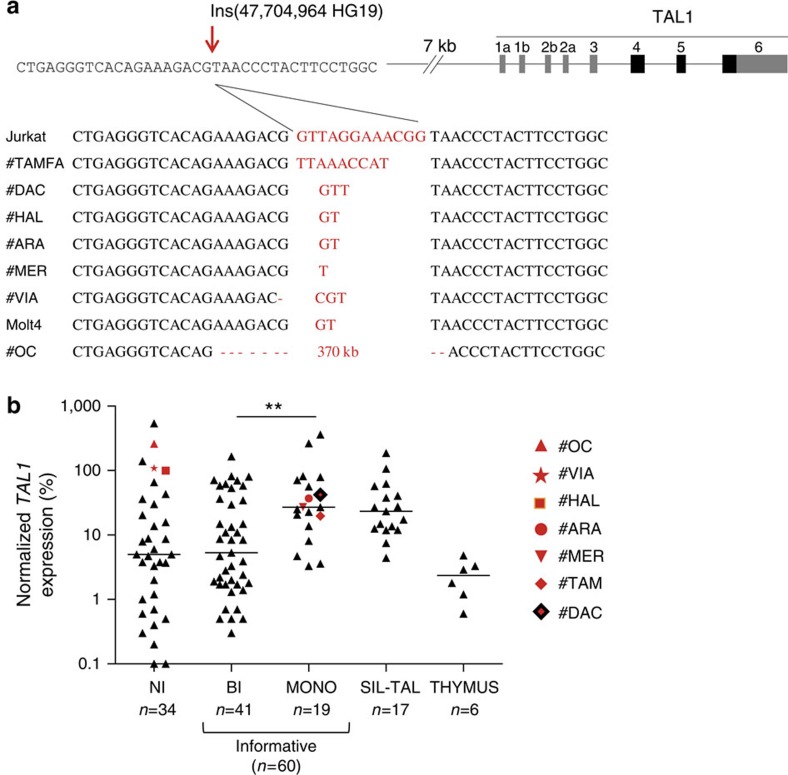
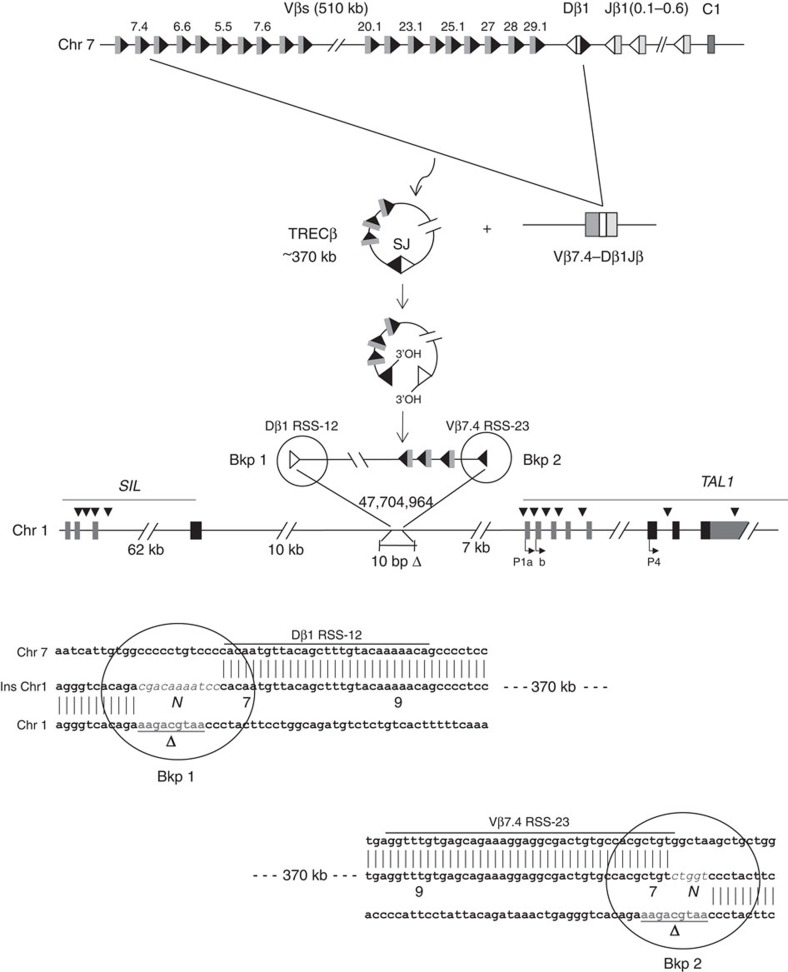
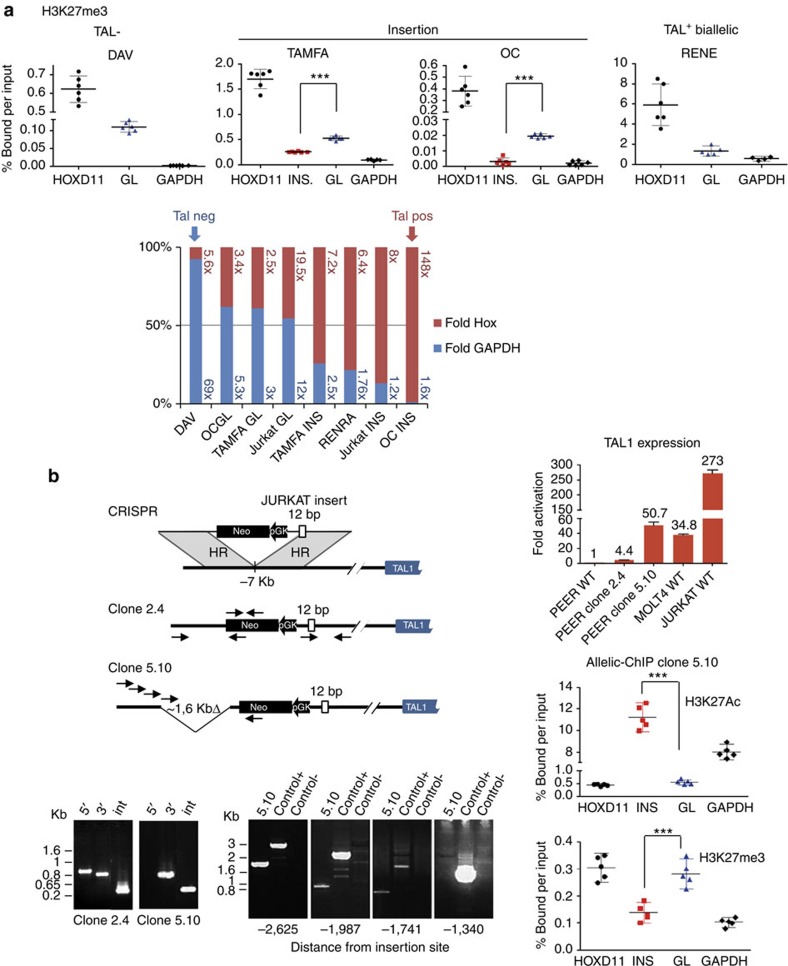
T-cell acute lymphoblastic leukaemias (T-ALL) are aggressive malignant proliferations characterized by high relapse rates and great genetic heterogeneity. TAL1 is amongst the most frequently deregulated oncogenes. Yet, over half of the TAL1(+) cases lack TAL1 lesions, suggesting unrecognized (epi)genetic deregulation mechanisms. Here we show that TAL1 is normally silenced in the T-cell lineage, and that the polycomb H3K27me3-repressive mark is focally diminished in TAL1(+) T-ALLs. Sequencing reveals that >20% of monoallelic TAL1(+) patients without previously known alterations display microinsertions or RAG1/2-mediated episomal reintegration in a single site 5' to TAL1. Using 'allelic-ChIP' and CrispR assays, we demonstrate that such insertions induce a selective switch from H3K27me3 to H3K27ac at the inserted but not the germline allele. We also show that, despite a considerable mechanistic diversity, the mode of oncogenic TAL1 activation, rather than expression levels, impact on clinical outcome. Altogether, these studies establish site-specific epigenetic desilencing as a mechanism of oncogenic activation.
T细胞急性淋巴细胞白血病(T-ALL)是侵袭性恶性增殖疾病,具有高复发率和高度遗传异质性。TAL1是最常失调的致癌基因之一。然而,超过一半的TAL1(+)病例缺乏TAL1病变,这表明存在未被认识的(表观)遗传失调机制。在这里,我们表明TAL1在T细胞谱系中通常是沉默的,并且在TAL1(+) T-ALL中多梳蛋白H3K27me3抑制标记局部减少。测序显示,超过20%的单等位基因TAL1(+)患者在之前没有已知改变的情况下,在TAL1上游5'端的单个位点显示微插入或RAG1/2介导的游离体重新整合。使用“等位基因ChIP”和CrispR分析,我们证明这种插入在插入的等位基因而非种系等位基因上诱导了从H3K27me3到H3K27ac的选择性转换。我们还表明,尽管存在相当大的机制多样性,但致癌性TAL1激活的模式而非表达水平会影响临床结果。总之,这些研究确立了位点特异性表观遗传去沉默作为致癌激活的一种机制。