Brown Alan, Long Fei, Nicholls Robert A, Toots Jaan, Emsley Paul, Murshudov Garib
MRC Laboratory of Molecular Biology, Francis Crick Avenue, Cambridge CB2 0QH, England.
Acta Crystallogr D Biol Crystallogr. 2015 Jan 1;71(Pt 1):136-53. doi: 10.1107/S1399004714021683.
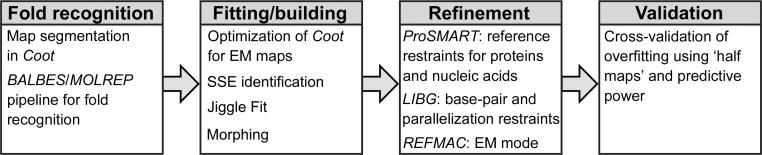
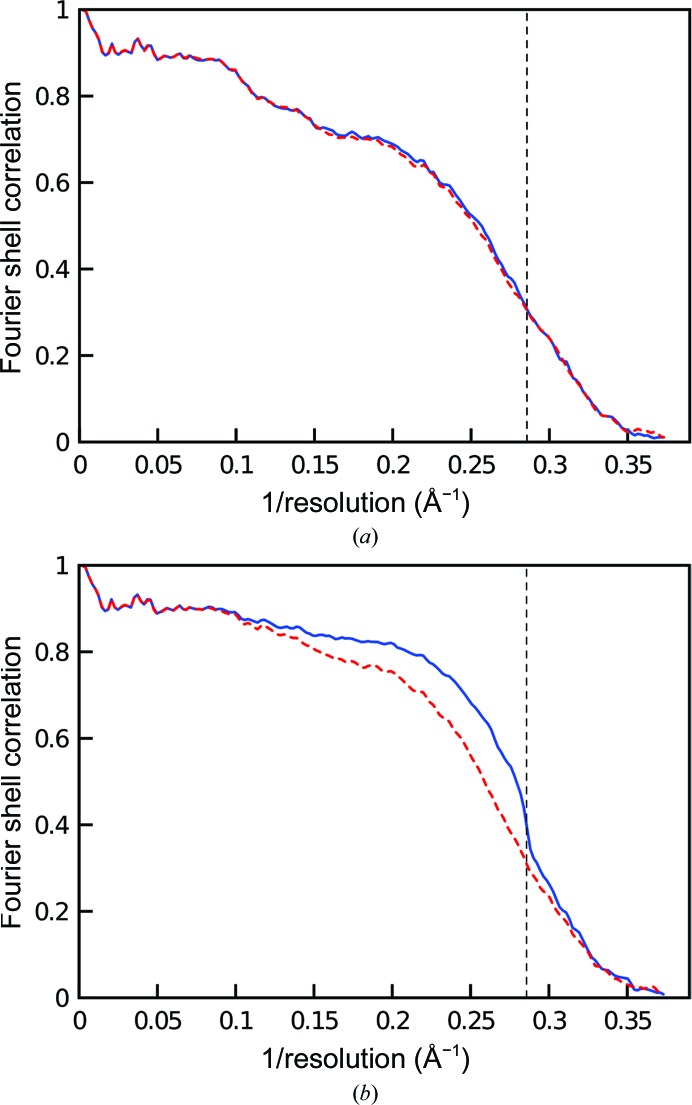
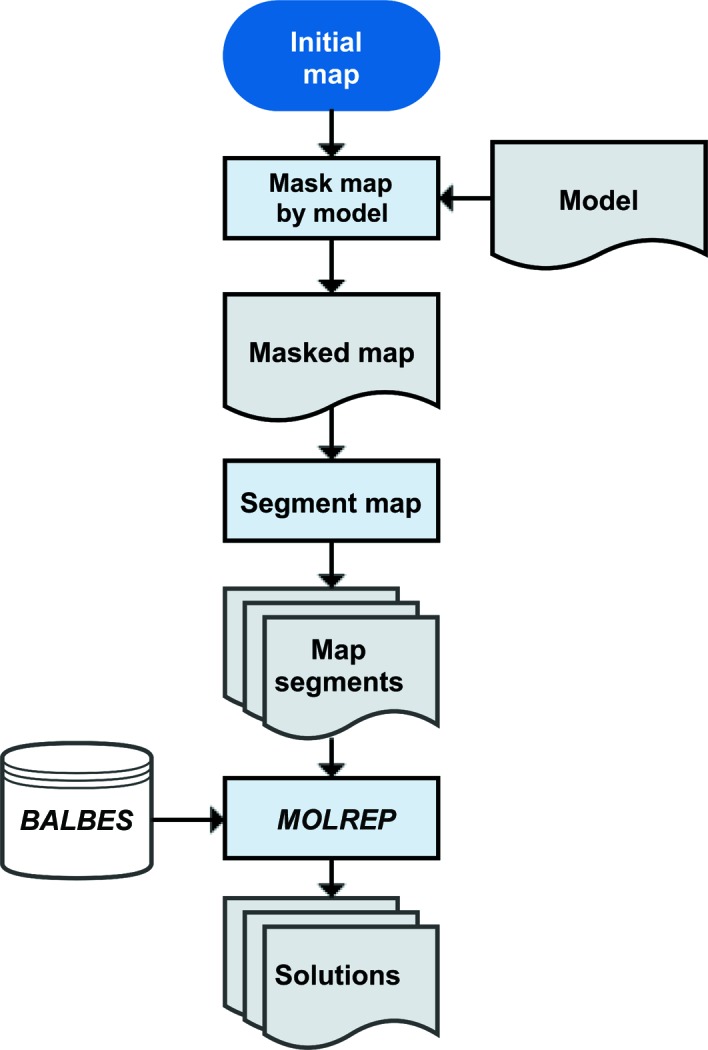
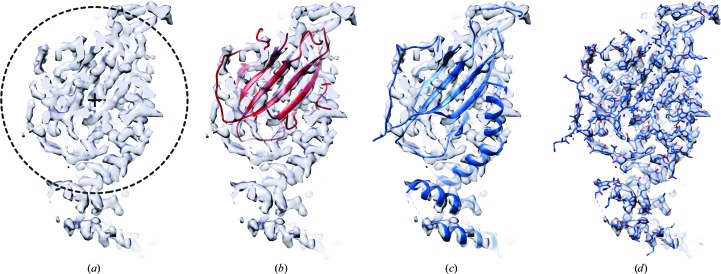
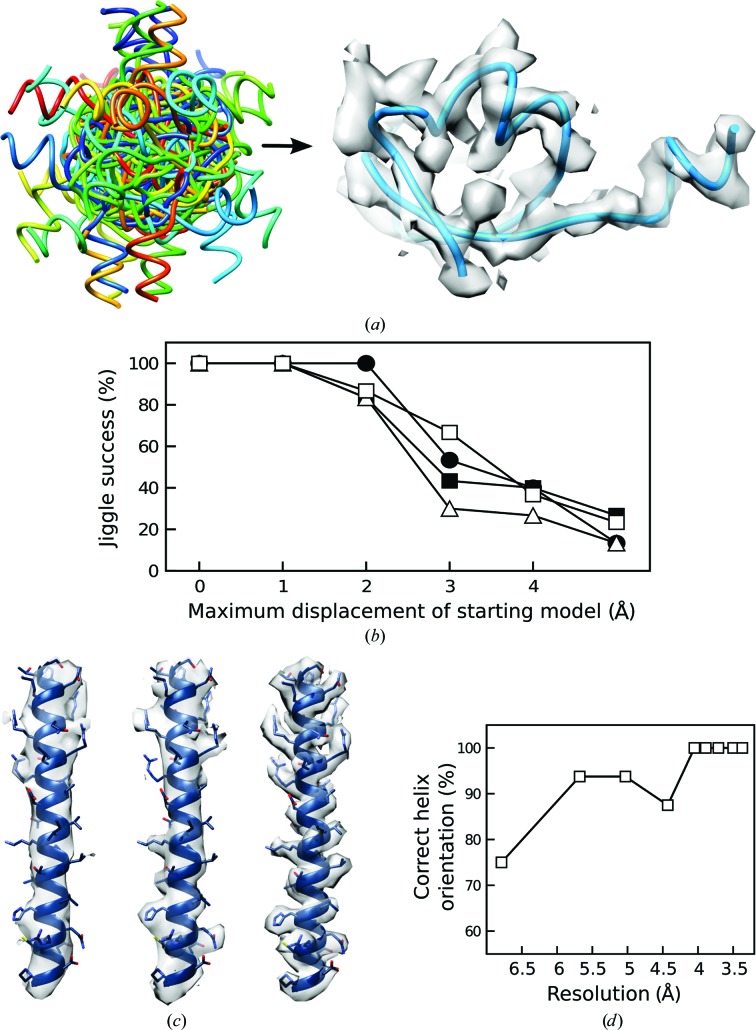
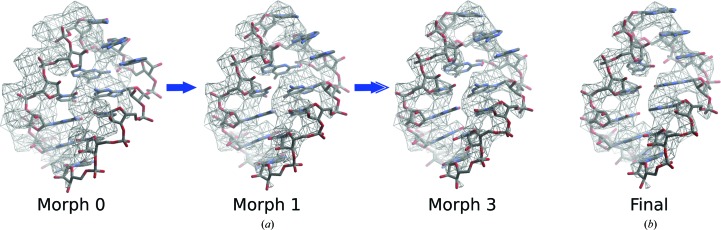
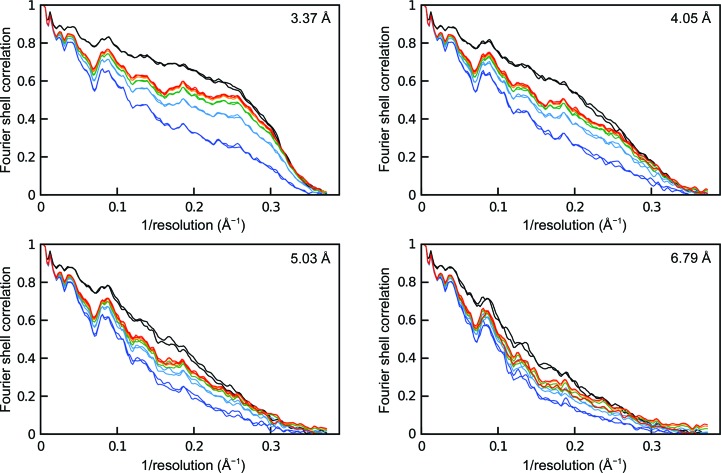
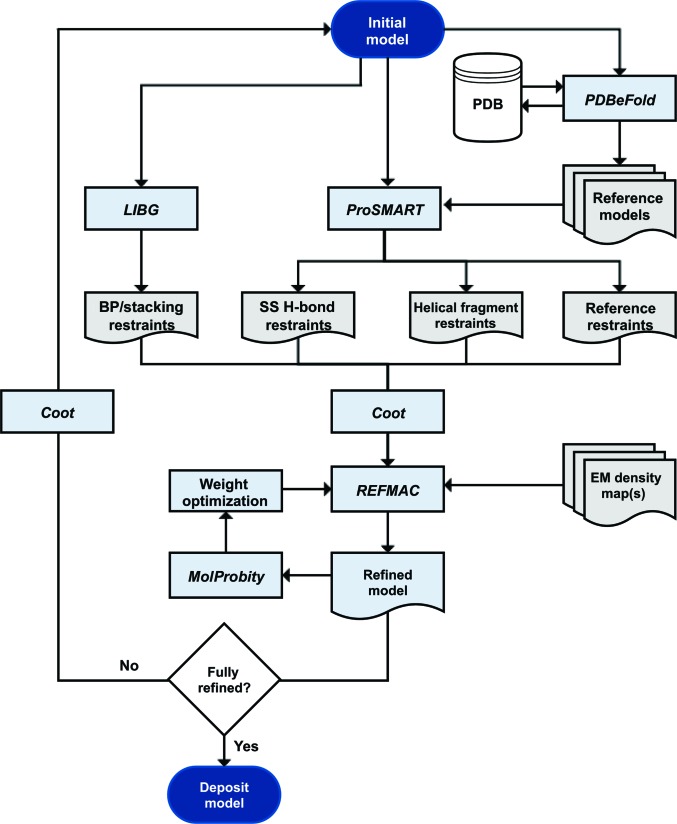
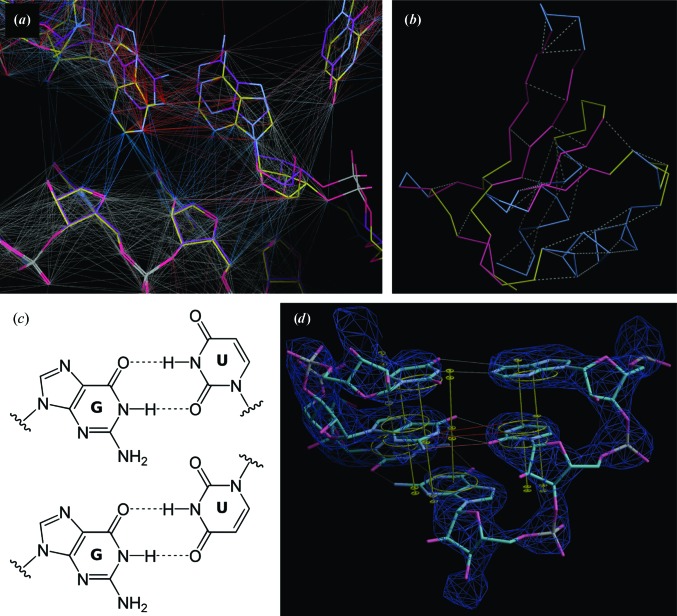
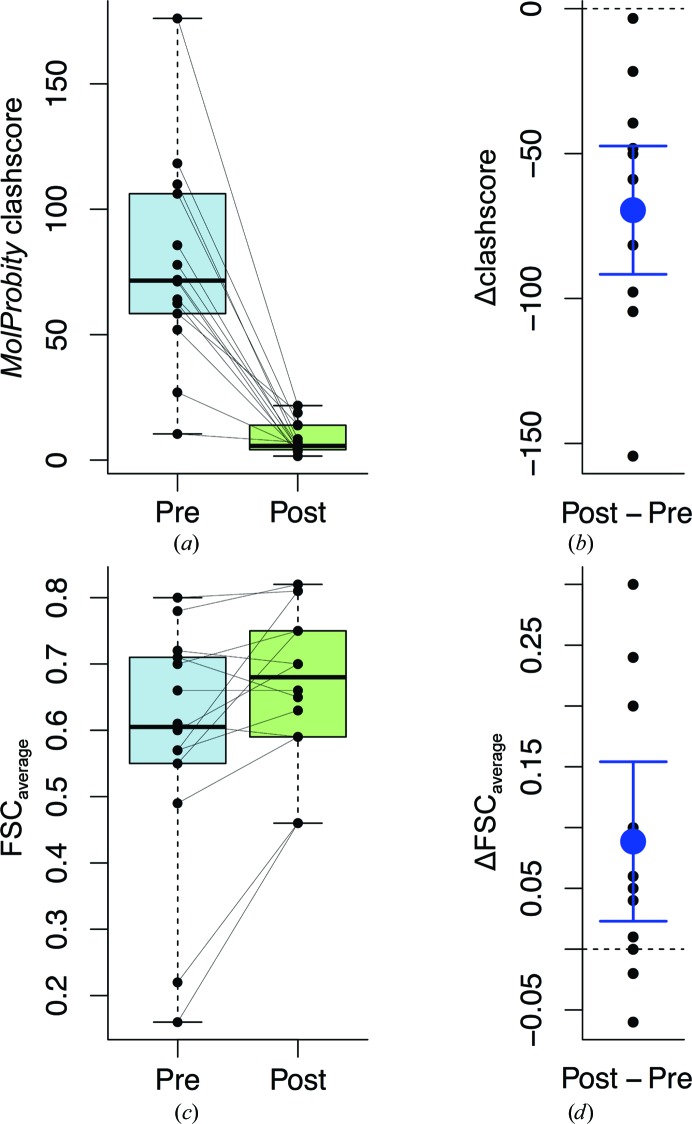
The recent rapid development of single-particle electron cryo-microscopy (cryo-EM) now allows structures to be solved by this method at resolutions close to 3 Å. Here, a number of tools to facilitate the interpretation of EM reconstructions with stereochemically reasonable all-atom models are described. The BALBES database has been repurposed as a tool for identifying protein folds from density maps. Modifications to Coot, including new Jiggle Fit and morphing tools and improved handling of nucleic acids, enhance its functionality for interpreting EM maps. REFMAC has been modified for optimal fitting of atomic models into EM maps. As external structural information can enhance the reliability of the derived atomic models, stabilize refinement and reduce overfitting, ProSMART has been extended to generate interatomic distance restraints from nucleic acid reference structures, and a new tool, LIBG, has been developed to generate nucleic acid base-pair and parallel-plane restraints. Furthermore, restraint generation has been integrated with visualization and editing in Coot, and these restraints have been applied to both real-space refinement in Coot and reciprocal-space refinement in REFMAC.
单颗粒冷冻电子显微镜技术(cryo-EM)最近的快速发展使得通过该方法能够在接近3埃的分辨率下解析结构。本文描述了一些有助于用立体化学合理的全原子模型解释电子显微镜重建结果的工具。BALBES数据库已被重新用作从密度图中识别蛋白质折叠的工具。对Coot的改进,包括新的抖动拟合和变形工具以及对核酸处理的改进,增强了其解释电子显微镜图的功能。REFMAC已被修改,以便将原子模型最佳拟合到电子显微镜图中。由于外部结构信息可以提高所推导原子模型的可靠性、稳定精修并减少过拟合,ProSMART已得到扩展,可从核酸参考结构生成原子间距离约束,并且已开发出一种新工具LIBG来生成核酸碱基对和平行平面约束。此外,约束生成已与Coot中的可视化和编辑集成,并且这些约束已应用于Coot中的实空间精修和REFMAC中的倒易空间精修。