Molecular Biophysics and Integrated Bioimaging Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
Cambridge Institute for Medical Research, University of Cambridge, Wellcome Trust/MRC Building, Hills Road, Cambridge CB2 0XY, England.
Acta Crystallogr D Struct Biol. 2018 Jun 1;74(Pt 6):531-544. doi: 10.1107/S2059798318006551. Epub 2018 May 30.
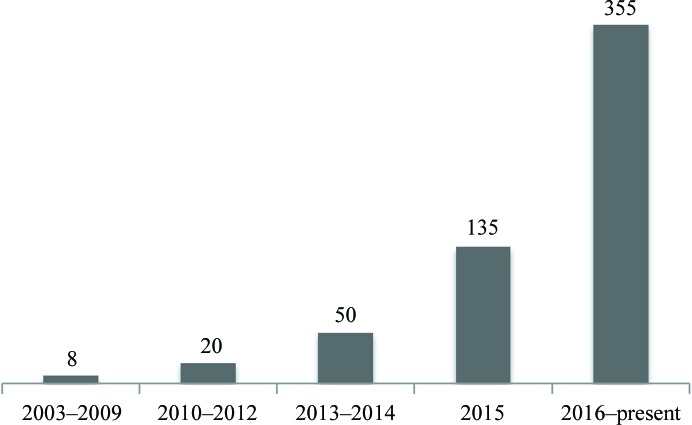
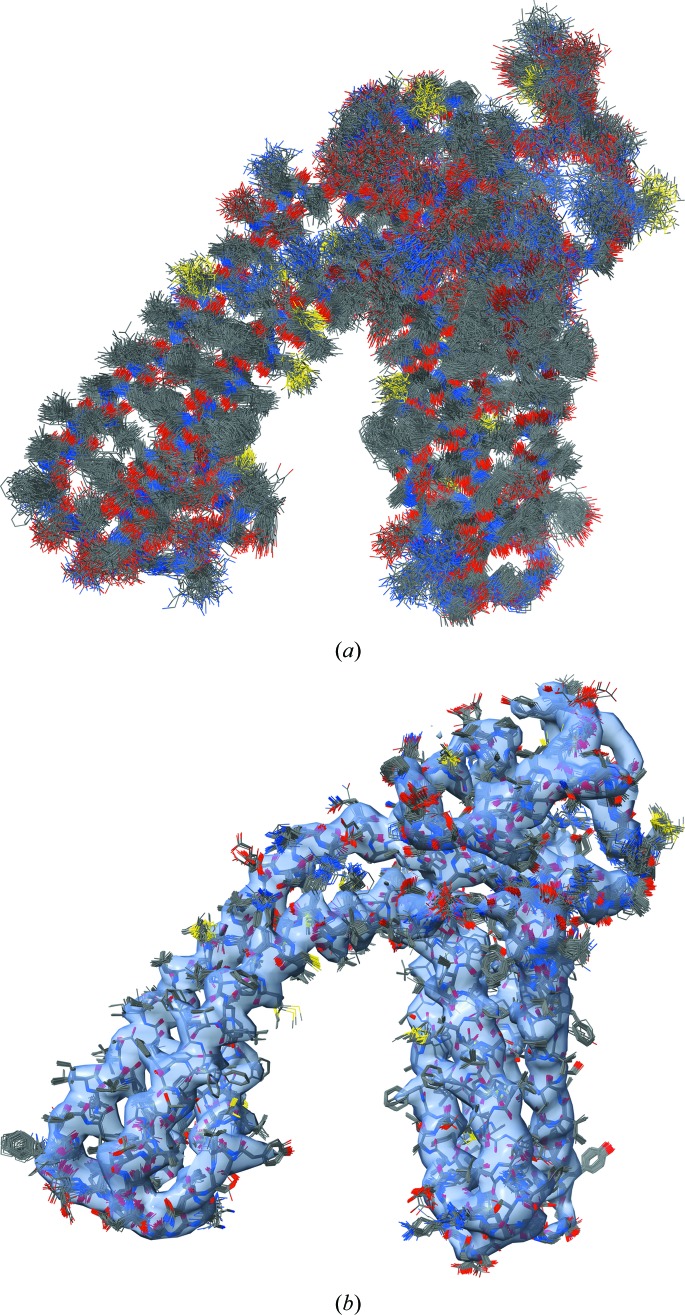
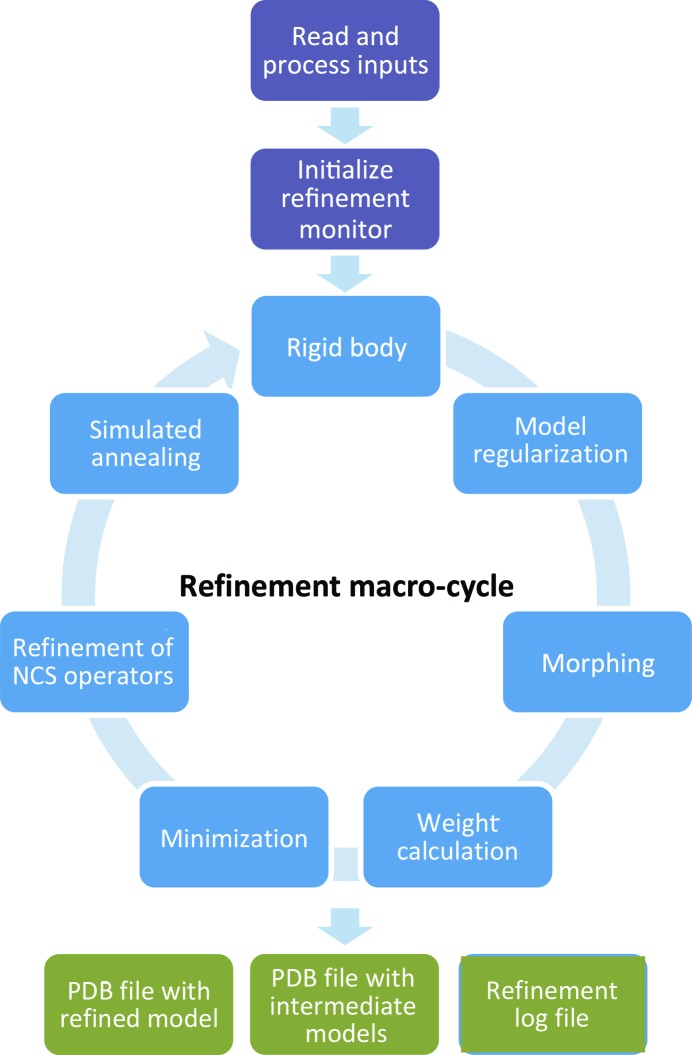
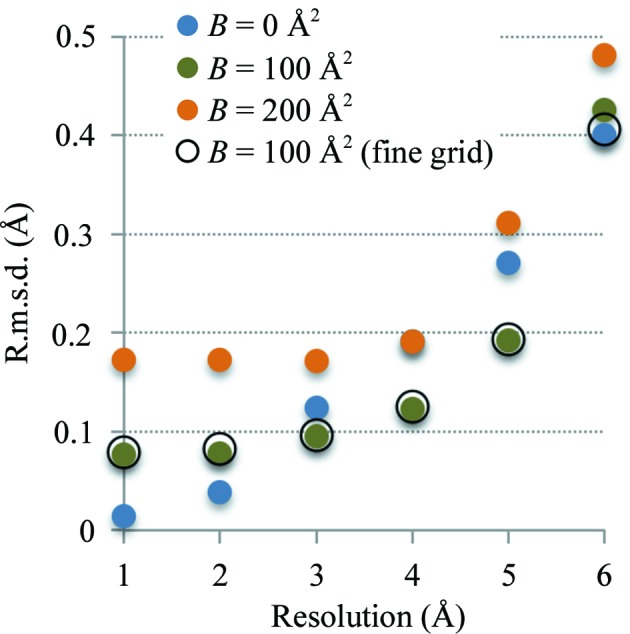
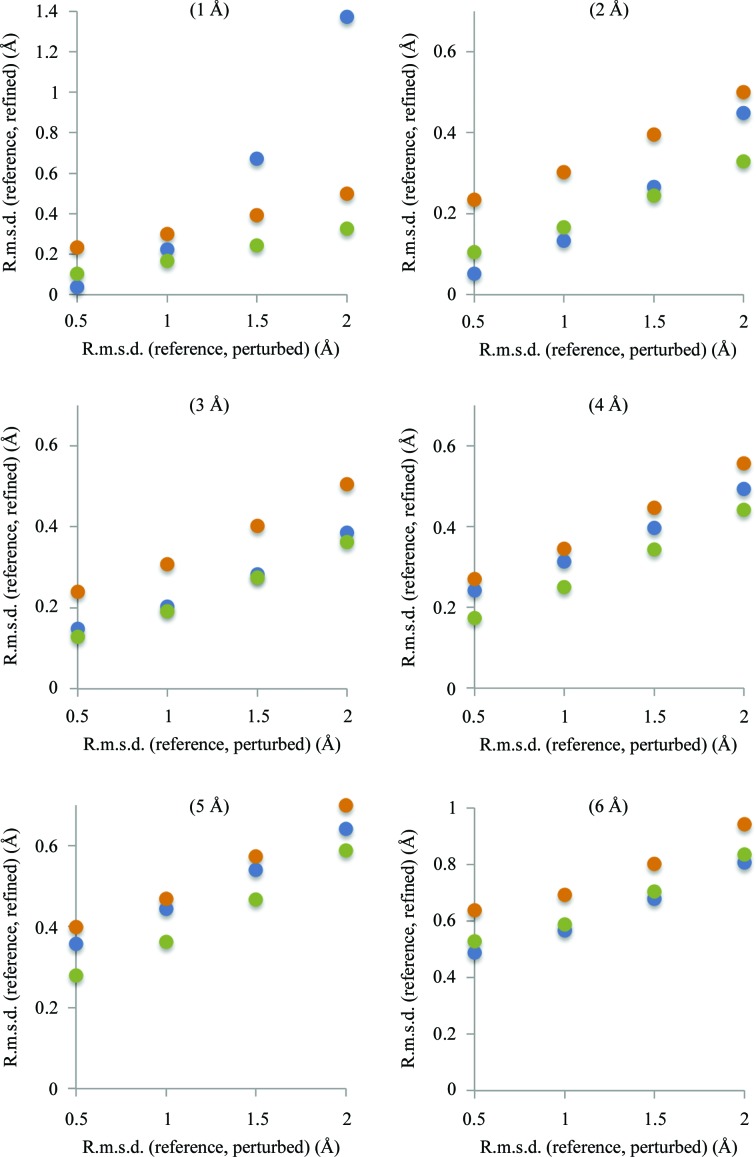
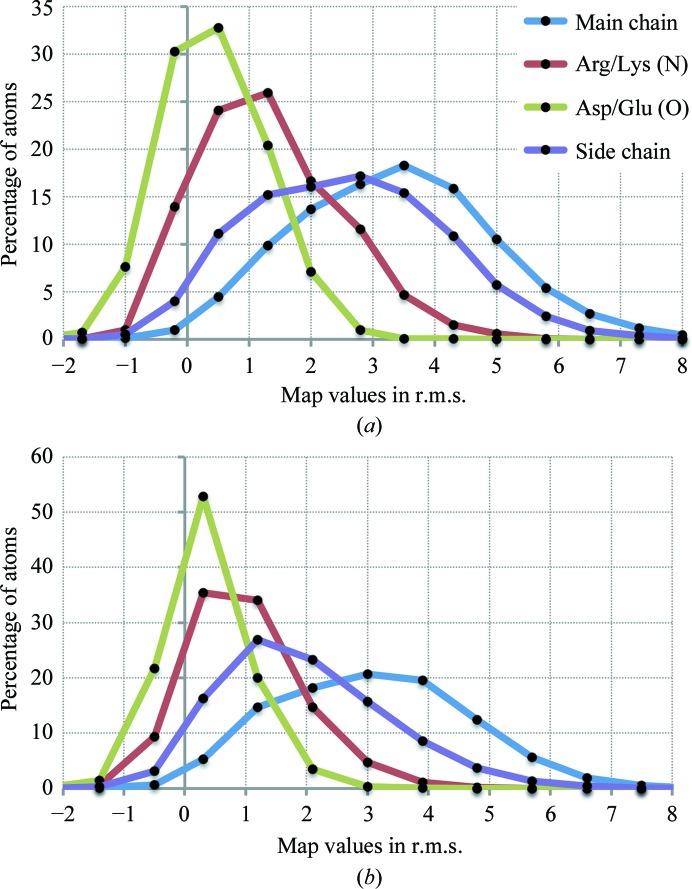
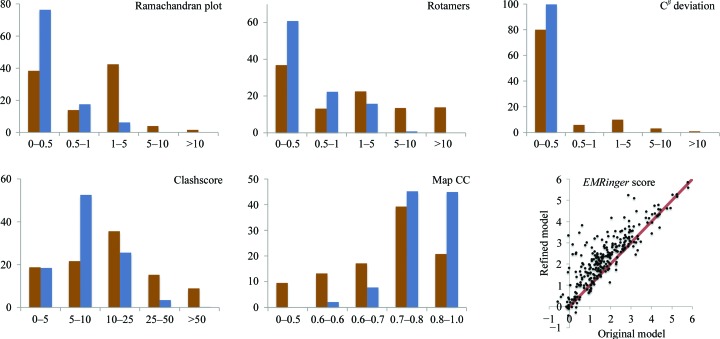
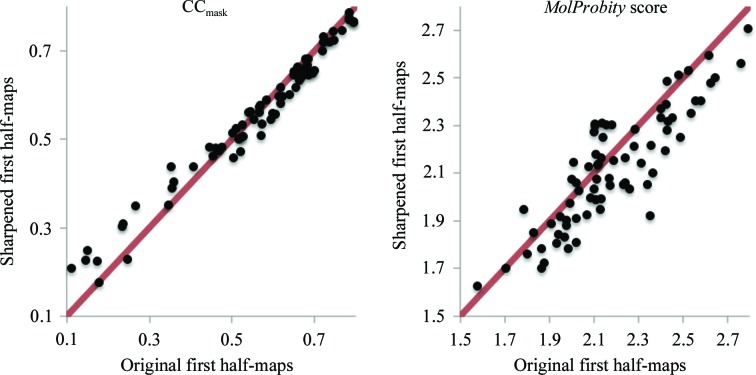
This article describes the implementation of real-space refinement in the phenix.real_space_refine program from the PHENIX suite. The use of a simplified refinement target function enables very fast calculation, which in turn makes it possible to identify optimal data-restraint weights as part of routine refinements with little runtime cost. Refinement of atomic models against low-resolution data benefits from the inclusion of as much additional information as is available. In addition to standard restraints on covalent geometry, phenix.real_space_refine makes use of extra information such as secondary-structure and rotamer-specific restraints, as well as restraints or constraints on internal molecular symmetry. The re-refinement of 385 cryo-EM-derived models available in the Protein Data Bank at resolutions of 6 Å or better shows significant improvement of the models and of the fit of these models to the target maps.
本文介绍了 PHENIX 套件中 phenix.real_space_refine 程序中实空间精修的实现。简化的精修目标函数的使用可以实现非常快速的计算,这反过来又使得可以在常规精修中识别最佳的数据约束权重,而几乎不会增加运行时间成本。针对低分辨率数据的原子模型精修可以受益于尽可能多的附加信息。除了对共价几何的标准约束外,phenix.real_space_refine 还利用了额外的信息,如二级结构和构象特异性约束,以及对内部分子对称性的约束或限制。对蛋白质数据库中可获得的 385 个分辨率在 6 Å 或更好的冷冻电镜衍生模型进行重新精修,显示出模型和这些模型与目标图的拟合都有显著改善。