Seiser Eric L, Innocenti Federico
Center for Pharmacogenomics and Individualized Therapy, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA.
Center for Pharmacogenomics and Individualized Therapy, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA. ; UNC Lineberger Comprehensive Cancer Center, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA.
Cancer Inform. 2015 Jan 27;13(Suppl 7):77-83. doi: 10.4137/CIN.S16345. eCollection 2014.
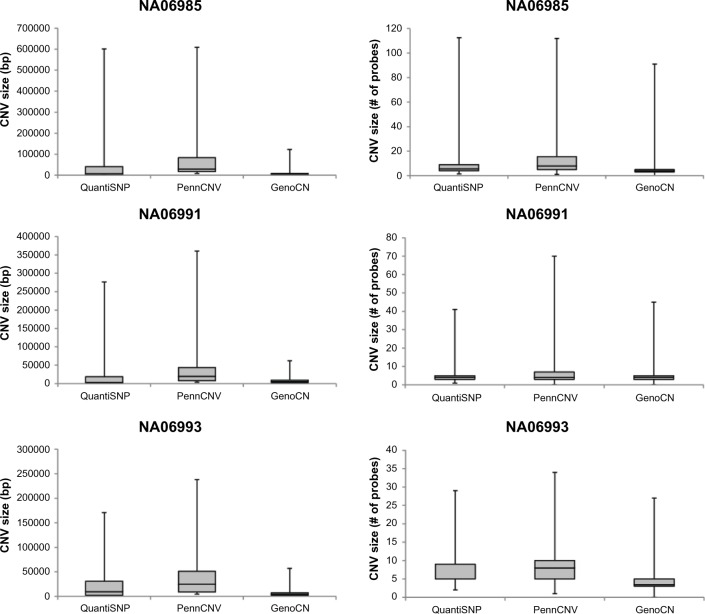
Somatic alterations in DNA copy number have been well studied in numerous malignancies, yet the role of germline DNA copy number variation in cancer is still emerging. Genotyping microarrays generate allele-specific signal intensities to determine genotype, but may also be used to infer DNA copy number using additional computational approaches. Numerous tools have been developed to analyze Illumina genotype microarray data for copy number variant (CNV) discovery, although commonly utilized algorithms freely available to the public employ approaches based upon the use of hidden Markov models (HMMs). QuantiSNP, PennCNV, and GenoCN utilize HMMs with six copy number states but vary in how transition and emission probabilities are calculated. Performance of these CNV detection algorithms has been shown to be variable between both genotyping platforms and data sets, although HMM approaches generally outperform other current methods. Low sensitivity is prevalent with HMM-based algorithms, suggesting the need for continued improvement in CNV detection methodologies.
DNA拷贝数的体细胞改变在众多恶性肿瘤中已得到充分研究,然而种系DNA拷贝数变异在癌症中的作用仍在不断显现。基因分型微阵列产生等位基因特异性信号强度以确定基因型,但也可使用额外的计算方法来推断DNA拷贝数。已经开发了许多工具来分析Illumina基因分型微阵列数据以发现拷贝数变异(CNV),尽管公众可免费使用的常用算法采用基于隐马尔可夫模型(HMM)的方法。QuantiSNP、PennCNV和GenoCN使用具有六个拷贝数状态的HMM,但在计算转移概率和发射概率的方式上有所不同。这些CNV检测算法的性能在基因分型平台和数据集之间已显示出差异,尽管基于HMM的方法通常优于其他现有方法。基于HMM的算法普遍存在低灵敏度的问题,这表明CNV检测方法需要持续改进。