Superti-Furga A, Pistone F, Romano C, Steinmann B
Department of Paediatrics, University of Zurich, Switzerland.
J Med Genet. 1989 Jun;26(6):358-62. doi: 10.1136/jmg.26.6.358.
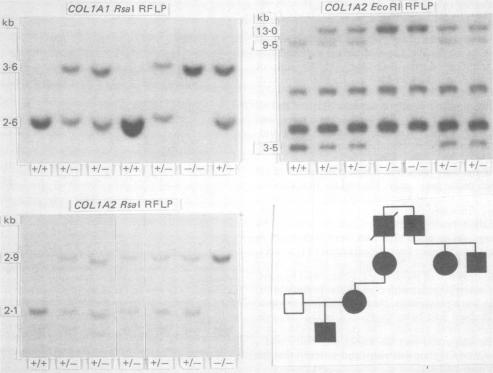
We report a family in which dominant osteogenesis imperfecta segregates with a COL1A2 haplotype and is associated with a structural defect in the helical region of the type I procollagen molecule. All affected subjects had short stature, dentinogenesis imperfecta, and myopia; however, great differences were observed in the number of fractures and in the degree of bone deformity. Identical biochemical changes were found in the type I collagen molecules synthesised by fibroblasts of subjects with severe or minimal bone fragility. These results confirm that mutations in the triple helical region of alpha 2(I) chains produce a milder phenotype than analogous mutations in the alpha 1(I) chains, but indicate that, in addition to defects in the type I collagen molecule, other factors may modulate the degree of bone involvement in osteogenesis imperfecta.
我们报告了一个家族,其中显性成骨不全与COL1A2单倍型共分离,并与I型前胶原分子螺旋区域的结构缺陷相关。所有受影响的个体均身材矮小、牙本质发育不全和近视;然而,在骨折数量和骨畸形程度方面观察到了很大差异。在骨脆性严重或轻微的受试者的成纤维细胞合成的I型胶原分子中发现了相同的生化变化。这些结果证实,α2(I)链三螺旋区域的突变产生的表型比α1(I)链中的类似突变更轻,但表明除了I型胶原分子的缺陷外,其他因素可能会调节成骨不全中骨受累的程度。