Jeong Seok Won, Chung Myungguen, Park Soo-Jung, Cho Seong Beom, Hong Kyung-Won
Division of Bio-Medical Informatics, Center for Genome Science, National Institute of Health, KCDC, Cheongju 363-951, Korea.
Genomics Inform. 2014 Dec;12(4):187-94. doi: 10.5808/GI.2014.12.4.187. Epub 2014 Dec 31.
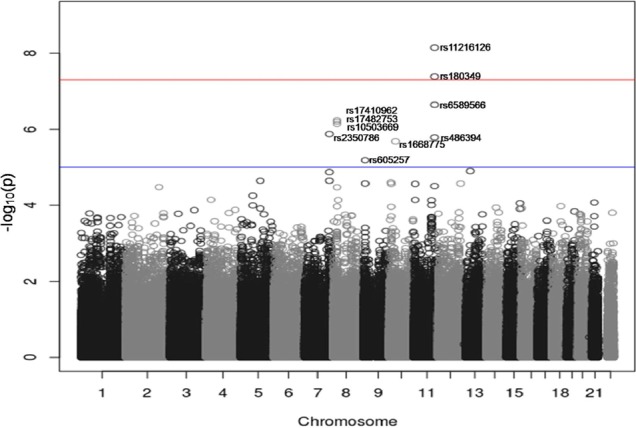
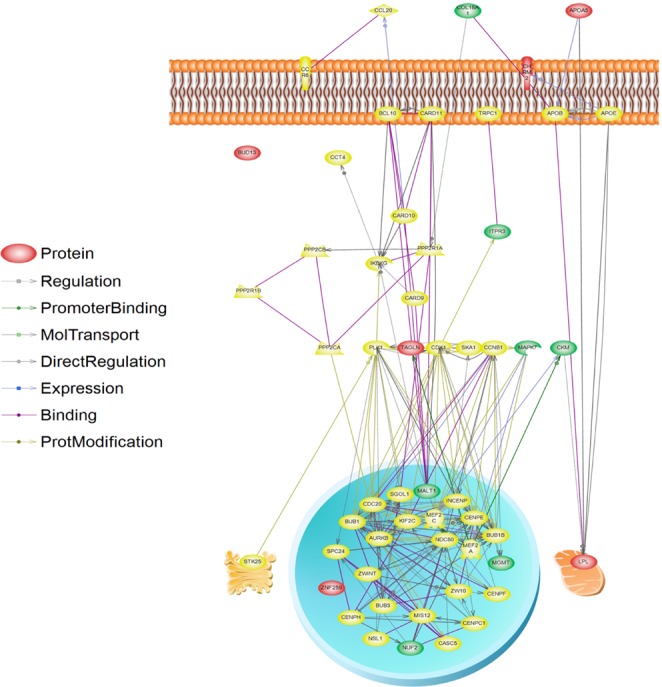
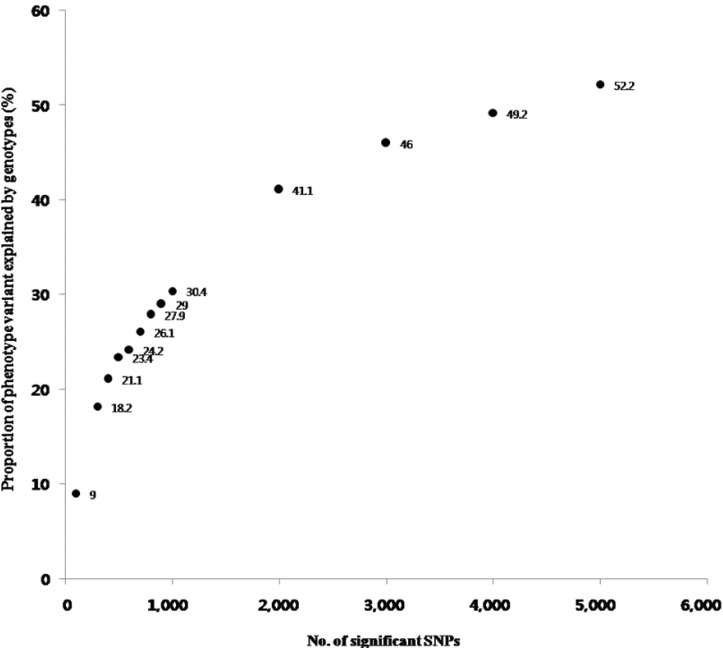
Metabolic syndrome (METS) is a disorder of energy utilization and storage and increases the risk of developing cardiovascular disease and diabetes. To identify the genetic risk factors of METS, we carried out a genome-wide association study (GWAS) for 2,657 cases and 5,917 controls in Korean populations. As a result, we could identify 2 single nucleotide polymorphisms (SNPs) with genome-wide significance level p-values (<5 × 10(-8)), 8 SNPs with genome-wide suggestive p-values (5 × 10(-8) ≤ p < 1 × 10(-5)), and 2 SNPs of more functional variants with borderline p-values (5 × 10(-5) ≤ p < 1 × 10(-4)). On the other hand, the multiple correction criteria of conventional GWASs exclude false-positive loci, but simultaneously, they discard many true-positive loci. To reconsider the discarded true-positive loci, we attempted to include the functional variants (nonsynonymous SNPs [nsSNPs] and expression quantitative trait loci [eQTL]) among the top 5,000 SNPs based on the proportion of phenotypic variance explained by genotypic variance. In total, 159 eQTLs and 18 nsSNPs were presented in the top 5,000 SNPs. Although they should be replicated in other independent populations, 6 eQTLs and 2 nsSNP loci were located in the molecular pathways of LPL, APOA5, and CHRM2, which were the significant or suggestive loci in the METS GWAS. Conclusively, our approach using the conventional GWAS, reconsidering functional variants and pathway-based interpretation, suggests a useful method to understand the GWAS results of complex traits and can be expanded in other genomewide association studies.
代谢综合征(METS)是一种能量利用和储存紊乱的病症,会增加患心血管疾病和糖尿病的风险。为了确定METS的遗传风险因素,我们在韩国人群中对2657例病例和5917例对照进行了全基因组关联研究(GWAS)。结果,我们能够鉴定出2个具有全基因组显著性水平p值(<5×10^(-8))的单核苷酸多态性(SNP),8个具有全基因组提示性p值(5×10^(-8)≤p<1×10^(-5))的SNP,以及2个具有临界p值(5×10^(-5)≤p<1×10^(-4))的更具功能变异的SNP。另一方面,传统GWAS的多重校正标准排除了假阳性位点,但同时也丢弃了许多真阳性位点。为了重新考虑被丢弃的真阳性位点,我们试图根据基因型方差解释的表型方差比例,将功能变异(非同义SNP [nsSNP]和表达数量性状位点 [eQTL])纳入前5000个SNP中。在前5000个SNP中总共呈现了159个eQTL和18个nsSNP。尽管它们应在其他独立人群中进行复制,但6个eQTL和2个nsSNP位点位于LPL、APOA5和CHRM2的分子途径中,这些是METS GWAS中的显著或提示性位点。总之,我们使用传统GWAS、重新考虑功能变异和基于途径的解释的方法,提出了一种理解复杂性状GWAS结果的有用方法,并且可以在其他全基因组关联研究中扩展。