Stöhr Anke C, López-Bueno Alberto, Blahak Silvia, Caeiro Maria F, Rosa Gonçalo M, Alves de Matos António Pedro, Martel An, Alejo Alí, Marschang Rachel E
Fachgebiet für Umwelt- und Tierhygiene, Universität Hohenheim, Stuttgart, Germany.
Centro de Biología Molecular Severo Ochoa (Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid), Madrid, Spain.
PLoS One. 2015 Feb 23;10(2):e0118633. doi: 10.1371/journal.pone.0118633. eCollection 2015.
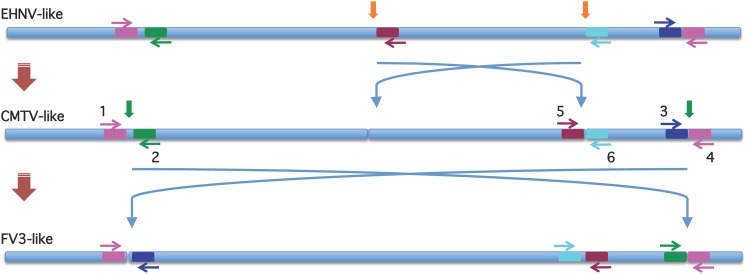
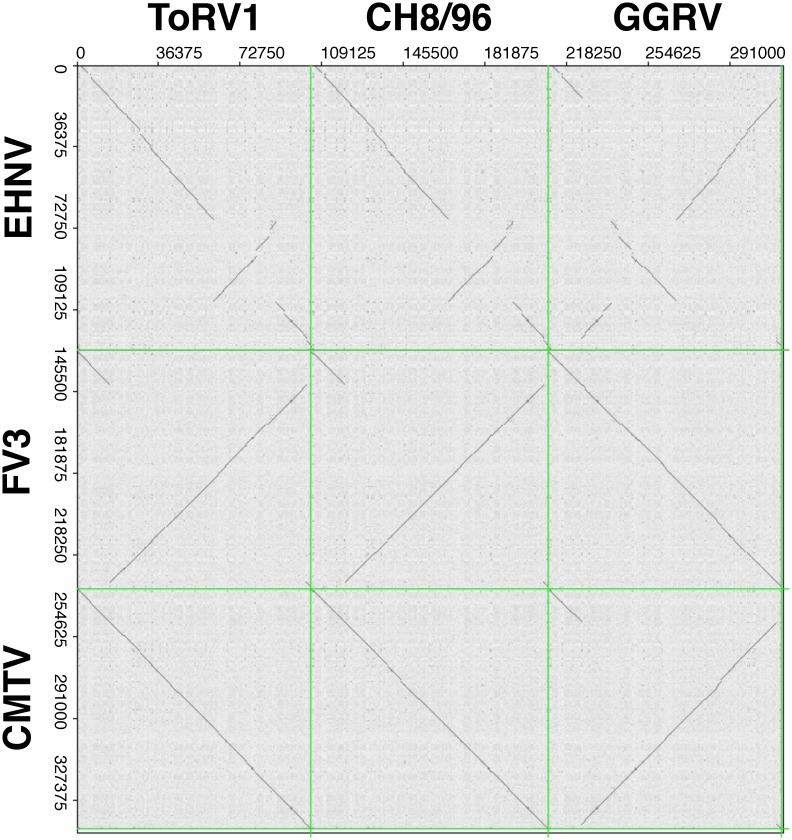
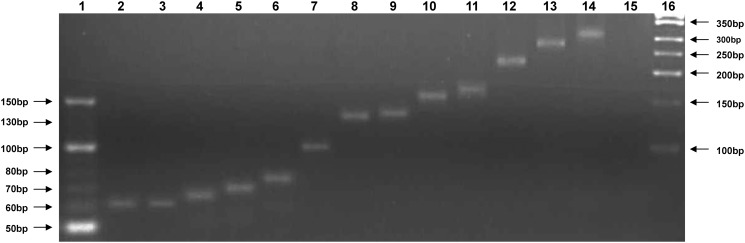
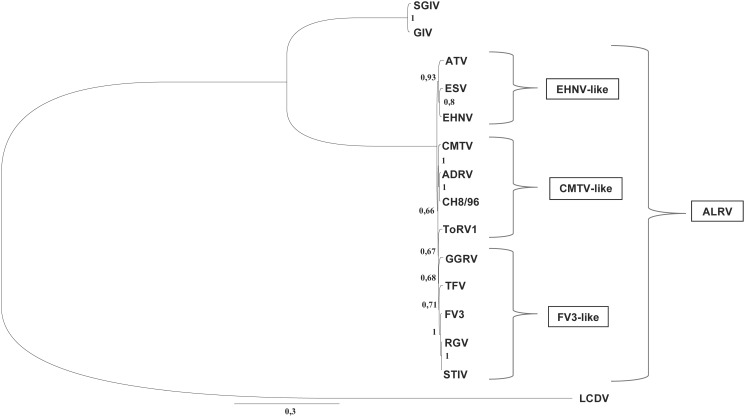
Ranaviruses in amphibians and fish are considered emerging pathogens and several isolates have been extensively characterized in different studies. Ranaviruses have also been detected in reptiles with increasing frequency, but the role of reptilian hosts is still unclear and only limited sequence data has been provided. In this study, we characterized a number of ranaviruses detected in wild and captive animals in Europe based on sequence data from six genomic regions (major capsid protein (MCP), DNA polymerase (DNApol), ribonucleoside diphosphate reductase alpha and beta subunit-like proteins (RNR-α and -β), viral homolog of the alpha subunit of eukaryotic initiation factor 2, eIF-2α (vIF-2α) genes and microsatellite region). A total of ten different isolates from reptiles (tortoises, lizards, and a snake) and four ranaviruses from amphibians (anurans, urodeles) were included in the study. Furthermore, the complete genome sequences of three reptilian isolates were determined and a new PCR for rapid classification of the different variants of the genomic arrangement was developed. All ranaviruses showed slight variations on the partial nucleotide sequences from the different genomic regions (92.6-100%). Some very similar isolates could be distinguished by the size of the band from the microsatellite region. Three of the lizard isolates had a truncated vIF-2α gene; the other ranaviruses had full-length genes. In the phylogenetic analyses of concatenated sequences from different genes (3223 nt/10287 aa), the reptilian ranaviruses were often more closely related to amphibian ranaviruses than to each other, and most clustered together with previously detected ranaviruses from the same geographic region of origin. Comparative analyses show that among the closely related amphibian-like ranaviruses (ALRVs) described to date, three recently split and independently evolving distinct genetic groups can be distinguished. These findings underline the wide host range of ranaviruses and the emergence of pathogen pollution via animal trade of ectothermic vertebrates.
两栖动物和鱼类中的蛙病毒被视为新兴病原体,在不同研究中已对多种分离株进行了广泛表征。在爬行动物中也越来越频繁地检测到蛙病毒,但爬行动物宿主的作用仍不清楚,仅提供了有限的序列数据。在本研究中,我们基于来自六个基因组区域(主要衣壳蛋白(MCP)、DNA聚合酶(DNApol)、核糖核苷二磷酸还原酶α和β亚基样蛋白(RNR-α和-β)、真核起始因子2α亚基的病毒同源物eIF-2α(vIF-2α)基因和微卫星区域)的序列数据,对在欧洲野生和圈养动物中检测到的多种蛙病毒进行了表征。该研究共纳入了来自爬行动物(乌龟、蜥蜴和一条蛇)的10种不同分离株以及来自两栖动物(无尾目、有尾目)的4种蛙病毒。此外,还确定了三种爬行动物分离株的完整基因组序列,并开发了一种新的PCR方法用于快速分类基因组排列的不同变体。所有蛙病毒在不同基因组区域的部分核苷酸序列上都有轻微差异(92.6 - 100%)。一些非常相似的分离株可以通过微卫星区域条带的大小来区分。蜥蜴分离株中有三个具有截短的vIF-2α基因;其他蛙病毒具有全长基因。在对不同基因的串联序列(3223 nt/10287 aa)进行系统发育分析时,爬行动物蛙病毒通常与两栖动物蛙病毒的亲缘关系比它们彼此之间的亲缘关系更近,并且大多数与先前从相同地理起源区域检测到的蛙病毒聚集在一起。比较分析表明,在迄今为止描述的密切相关的两栖动物样蛙病毒(ALRVs)中,可以区分出三个最近分裂且独立进化的不同遗传组。这些发现强调了蛙病毒广泛的宿主范围以及通过变温脊椎动物的动物贸易出现的病原体污染问题。