Paemka Lily, Mahajan Vinit B, Ehaideb Salleh N, Skeie Jessica M, Tan Men Chee, Wu Shu, Cox Allison J, Sowers Levi P, Gecz Jozef, Jolly Lachlan, Ferguson Polly J, Darbro Benjamin, Schneider Amy, Scheffer Ingrid E, Carvill Gemma L, Mefford Heather C, El-Shanti Hatem, Wood Stephen A, Manak J Robert, Bassuk Alexander G
Department of Pediatrics, The University of Iowa, Iowa City, Iowa, United States of America; Interdisciplinary Program in Genetics, The University of Iowa, Iowa City, Iowa, United States of America.
Department of Ophthalmology and Visual Sciences, The University of Iowa, Iowa City, Iowa, United States of America; Roy J. and Lucille A. Carver College of Medicine, The University of Iowa, Iowa City, Iowa, United States of America; Department of Biology, The University of Iowa, Iowa City, Iowa, United States of America.
PLoS Genet. 2015 Mar 12;11(3):e1005022. doi: 10.1371/journal.pgen.1005022. eCollection 2015 Mar.
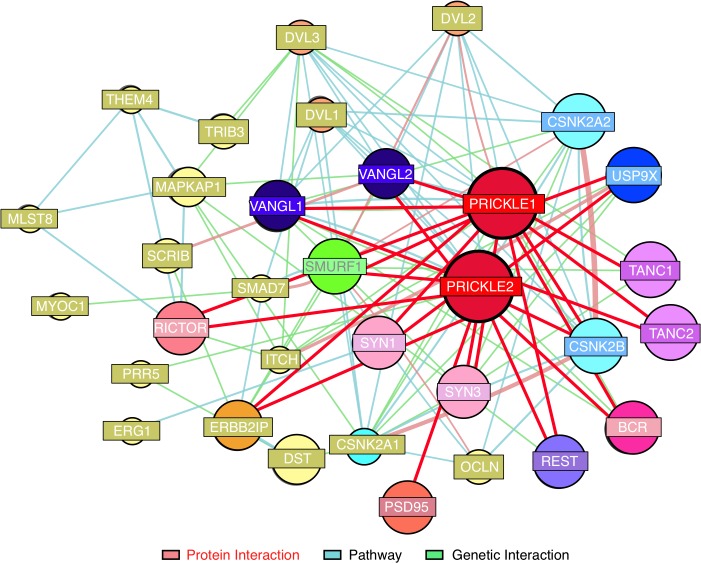
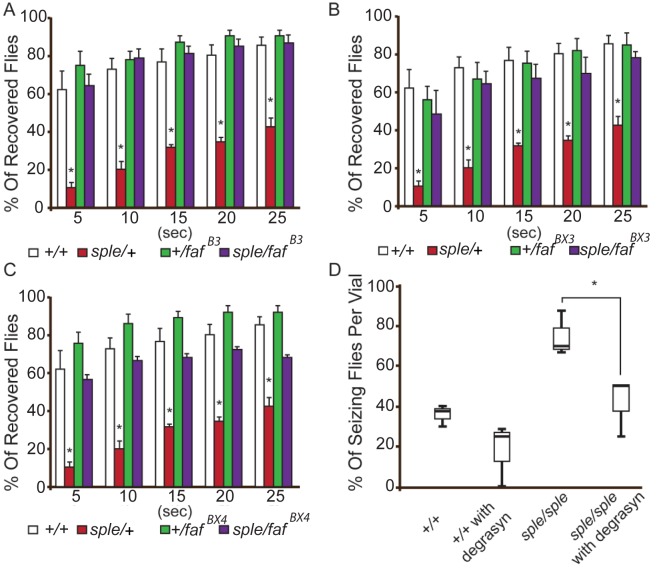
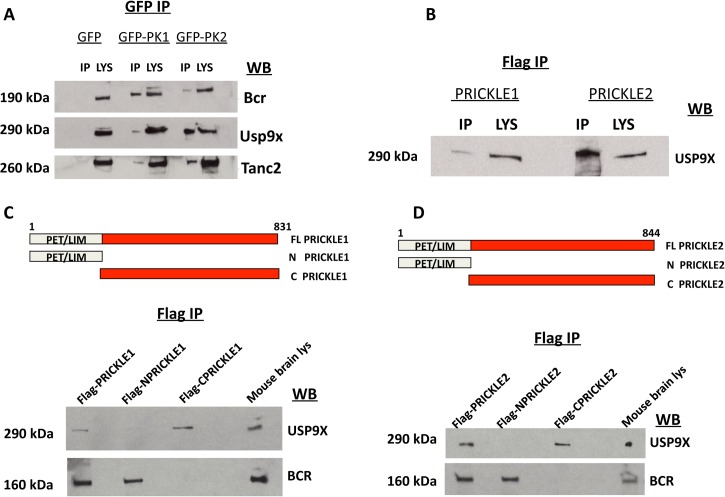
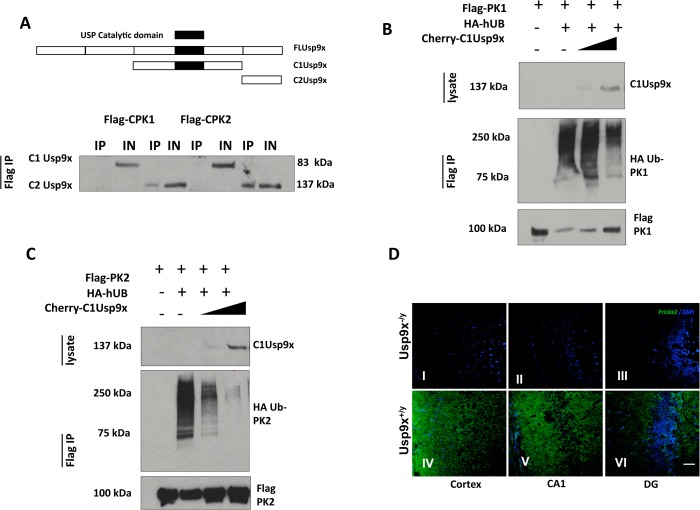
Epilepsy is a common disabling disease with complex, multifactorial genetic and environmental etiology. The small fraction of epilepsies subject to Mendelian inheritance offers key insight into epilepsy disease mechanisms; and pathologies brought on by mutations in a single gene can point the way to generalizable therapeutic strategies. Mutations in the PRICKLE genes can cause seizures in humans, zebrafish, mice, and flies, suggesting the seizure-suppression pathway is evolutionarily conserved. This pathway has never been targeted for novel anti-seizure treatments. Here, the mammalian PRICKLE-interactome was defined, identifying prickle-interacting proteins that localize to synapses and a novel interacting partner, USP9X, a substrate-specific de-ubiquitinase. PRICKLE and USP9X interact through their carboxy-termini; and USP9X de-ubiquitinates PRICKLE, protecting it from proteasomal degradation. In forebrain neurons of mice, USP9X deficiency reduced levels of Prickle2 protein. Genetic analysis suggests the same pathway regulates Prickle-mediated seizures. The seizure phenotype was suppressed in prickle mutant flies by the small-molecule USP9X inhibitor, Degrasyn/WP1130, or by reducing the dose of fat facets a USP9X orthologue. USP9X mutations were identified by resequencing a cohort of patients with epileptic encephalopathy, one patient harbored a de novo missense mutation and another a novel coding mutation. Both USP9X variants were outside the PRICKLE-interacting domain. These findings demonstrate that USP9X inhibition can suppress prickle-mediated seizure activity, and that USP9X variants may predispose to seizures. These studies point to a new target for anti-seizure therapy and illustrate the translational power of studying diseases in species across the evolutionary spectrum.
癫痫是一种常见的致残性疾病,其病因复杂,涉及多因素的遗传和环境因素。一小部分遵循孟德尔遗传规律的癫痫为深入了解癫痫发病机制提供了关键线索;单基因突变引发的病理变化可为通用治疗策略指明方向。PRICKLE基因的突变可在人类、斑马鱼、小鼠和果蝇中引发癫痫发作,这表明癫痫抑制通路在进化上是保守的。该通路从未被作为新型抗癫痫治疗的靶点。在此,我们确定了哺乳动物PRICKLE相互作用组,鉴定出定位于突触的与PRICKLE相互作用的蛋白以及一个新的相互作用伴侣USP9X,一种底物特异性去泛素化酶。PRICKLE和USP9X通过它们的羧基末端相互作用;USP9X使PRICKLE去泛素化,保护其免受蛋白酶体降解。在小鼠前脑神经元中,USP9X缺乏会降低Prickle2蛋白的水平。遗传分析表明,相同的通路调节PRICKLE介导的癫痫发作。在prickle突变果蝇中,小分子USP9X抑制剂Degrasyn/WP1130或降低USP9X同源物fat facets的剂量可抑制癫痫表型。通过对一组癫痫性脑病患者进行重测序鉴定出USP9X突变,一名患者携带一个新生错义突变,另一名患者携带一个新的编码突变。这两个USP9X变体均位于与PRICKLE相互作用的结构域之外。这些发现表明,抑制USP9X可抑制PRICKLE介导的癫痫发作活动,且USP9X变体可能易引发癫痫发作。这些研究指出了一个新的抗癫痫治疗靶点,并说明了在整个进化谱系的物种中研究疾病的转化潜力。